
Go to the page with the list of problems.
Aromatic Substitution Reactions in Benzene and Benzene Derivatives – solutions to problems
Solution 1:
Assuming that all the structures shown are planar, cyclic and conjugated, the next condition to be fulfilled for them to be aromatic is Hückel’s rule (4n+2) i.e. they must present 2,6,10,14,18, etc.. electrons π in the conjugated system to be aromatic and (4n) 4, 8, 12, 16, etc.. to be antiaromatic. Therefore, structures II (cycloheptatrienyl cation), III (azulene), IV (cyclopropenyl cation), VI (trans-15,16-dihydropyrene), VII ([18]anulene), VIII (anthracene), IX (cyclooctatetraene dianion), X ([16]anulene dication), XI (pyridine) and XII (pyrrole) will be aromatic. They will be antiaromatic, structures I ([16]anulene) and V (S-indacene).
Solution 2:
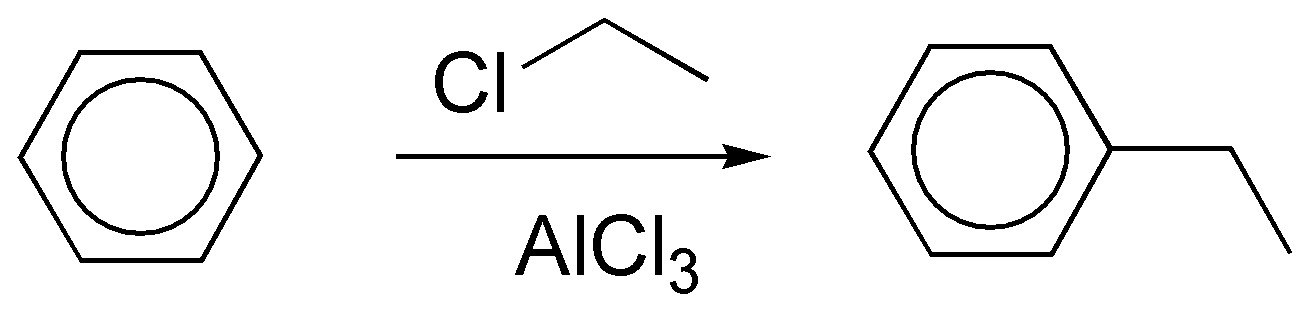
(a) It is prepared by conventional Friedel-Crafts alkylation. Moreover, there is no possibility of transpositions because the starting ethyl chloride generates only one carbocation.
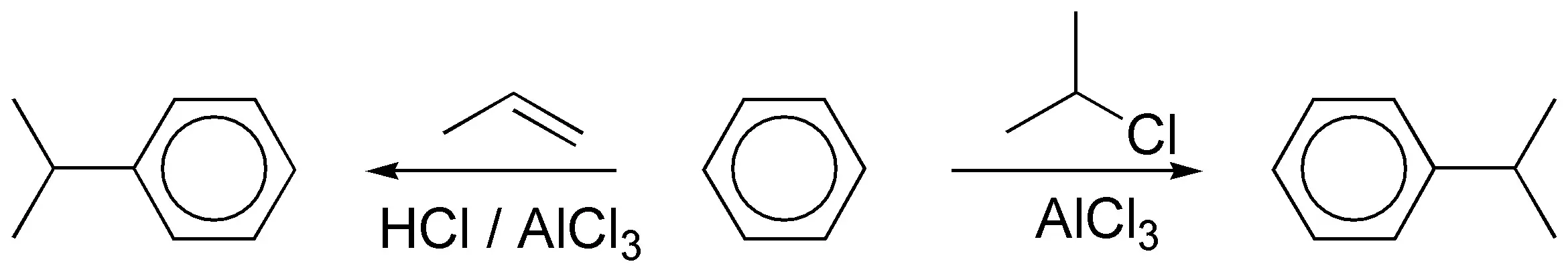
b) It can be prepared from propene with acid catalysis to generate a secondary carbocation, which acts as a nucleophile, or from the secondary alkyl halide.
c) The alkylation of an arene with a primary alkyl halide with more than two carbon atoms in the presence of a catalyst does not lead to the formation of the expected product, since mixtures are produced, as a consequence of the transposition of the intermediate primary carbocation postulated in the reaction. In this case it is necessary to proceed to acylation with an acyl chloride whose carbon skeleton matches that of the radical to be introduced, to give a ketone, which can then be reduced.
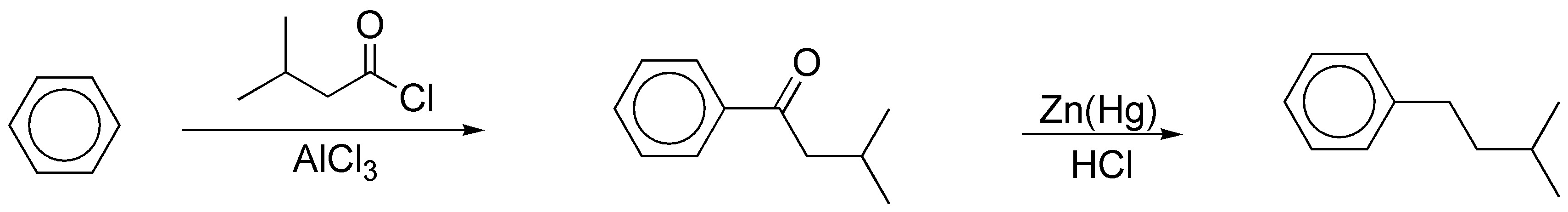
The reduction described is the application of the Clemmensen reaction, although the Wolff-Kishner reaction (NH2NH2 / KOH / ethylene glycol) could also have been used, since in the substrate there are no groups incompatible with the acid medium of the Clemmensen reaction nor with the base used in the Wolff-Kishner reaction.
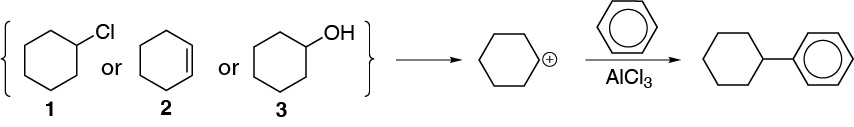
d) The proposed product can be obtained by three procedures:
- From cyclohexyl chloride with a Lewis acid.
- From cyclohexene with catalysis of a mineral acid.
- From cyclohexanol with catalysis of a mineral acid.
In all three cases a secondary carbocation is formed which acts as an electrophile.
Solution 3:
a) By Friedel-Crafts alkylation with benzyl bromide the compound can be prepared according to the scheme:
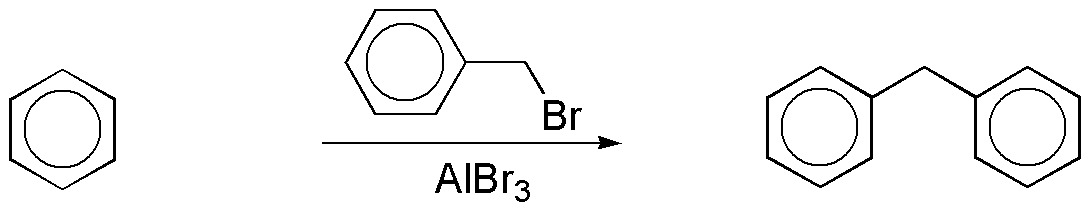
In turn, benzyl bromide can be obtained from benzene in two steps:
Note that a slight excess (1.1 mol) has been used to avoid the formation of polysubstituted products, since the aromatic ring of toluene is richer in electrons than benzene itself, given the activating character of the methyl group.
b) The synthesis of the proposed bicyclic compound can be carried out directly by means of a Friedel-Crafts acylation on benzene.
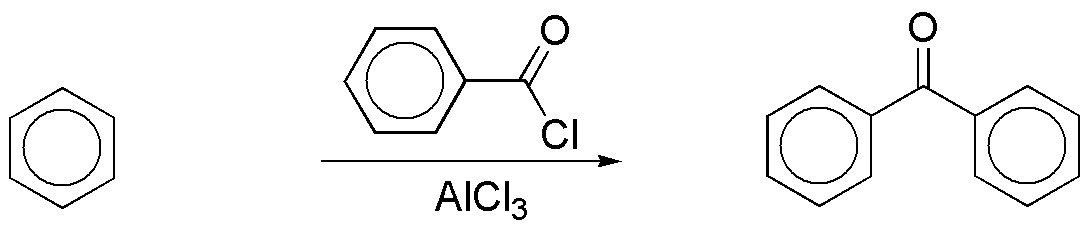
To obtain the benzoyl chloride used in the reaction, the following sequence of reactions can be followed:
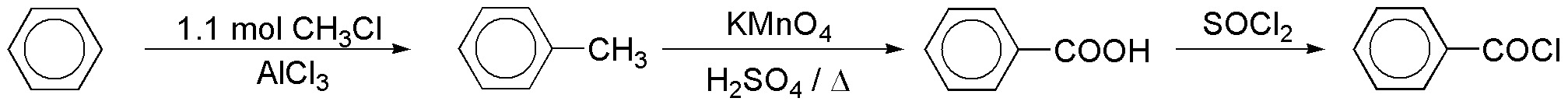
Benzene is transformed into toluene, and the latter into benzoyl acid by oxidation with permanganate in an acid medium. Benzoyl chloride is prepared by reaction of the acid with thionyl chloride.
c) In biphenyl a direct C-C bond is established between two benzene rings. A possible synthesis of biphenyl can be made from bromobenzene.
The bromobenzene is converted to magnesian which reacts with another bromobenzene molecule, to give biphenyl.

Solution 4:
The rate of electrophilic aromatic substitution reaction depends on whether the group increases or decreases the electron density on the aromatic ring. The reaction rate of benzene is taken as a reference. We can classify the compounds into:
I: moderate activator, II: weak deactivator, III: strong deactivator, IV: moderate activator, V: weak deactivator, VI: strong deactivator, VII: moderate deactivator and VIII: strong deactivator.
and the reaction rate order will be as indicated in the figure:
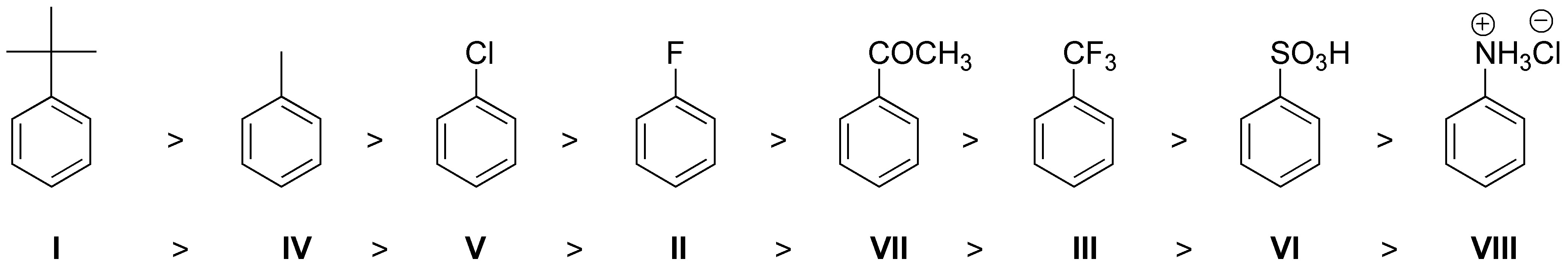
The reaction rate of I should be higher than that of IV, since the tert-butyl group is richer in electrons than methyl and presents greater inductive effect +I. In addition, fluorine being more electronegative, is more deactivating than chlorine, so V has a higher reaction rate than II.
Solution 5:
Aniline cannot be nitrated directly, since nitric acid oxidizes the amino group, so it is necessary to protect the amino group by forming a derivative, which subsequently allows the -NH2 to be regenerated relatively easily. For this purpose, the aniline is treated with acetic anhydride to obtain the acetanilide, which is nitrated under conventional conditions. Subsequently, the protective group is removed by acid hydrolysis to obtain the desired product.

Note that in the nitration reaction the monosubstituted para derivative is obtained as the majority.
This result is justified for two reasons:
- The acetamido group buffers the activating character of -NH2, thus minimizing polysubstitution.
- Hardly any significant amount of the product is obtained at ortho-, since this position from the steric point of view is less favored by the bulk of the acetamido group.
Solution 6:
a) To introduce a nitro group and a bromine atom into the benzene ring we will have to perform two consecutive electrophilic aromatic substitution reactions. The sequence of reactions is key, since the nitro group is meta-directing and the bromine ortho– and para-directing. If nitration proceeds first the second substituent will inevitably enter meta-, whereas if halogenation occurs. The two possible options are:
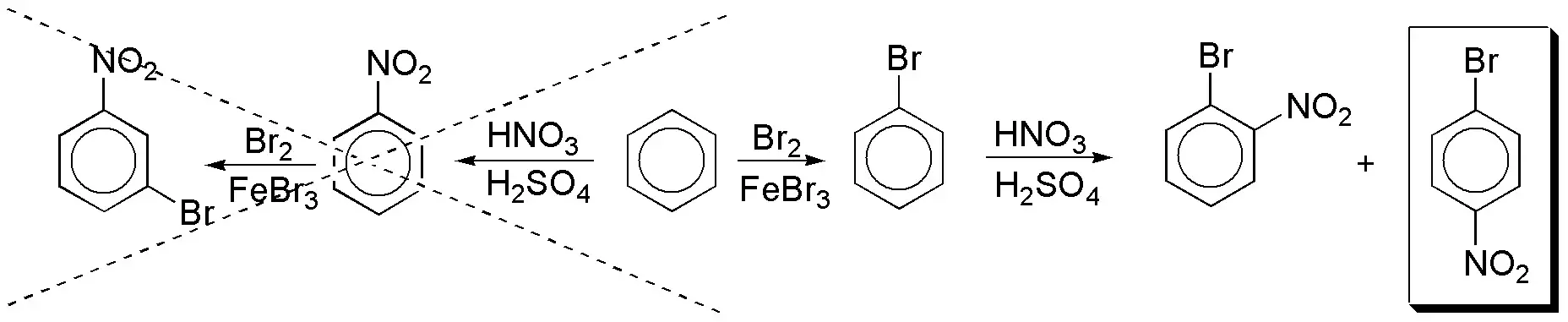
In the correct sequence of reactions, the desired product is obtained accompanied by the ortho– isomer, so if you want to obtain pure, you will have to proceed to separate them.
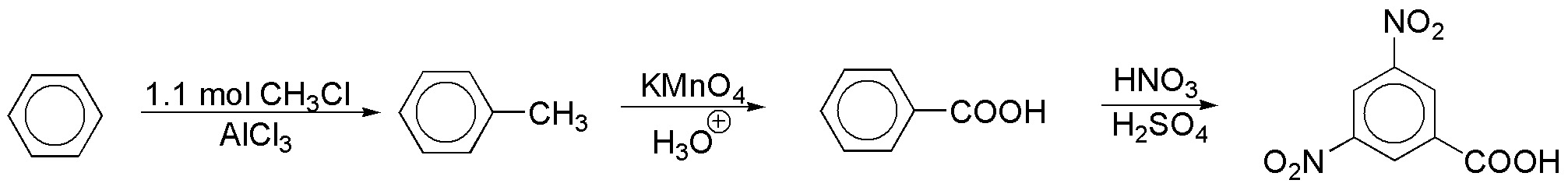
b) All groups are meta-directive. However, the COOH group is less deactivating than the nitro group, so it should be introduced earlier to facilitate, as far as possible, the formation of the desired product. As is known, the COOH group can be obtained from an alkylbenzene by oxidation with KMnO4 or K2Cr2O7 in an acid medium.
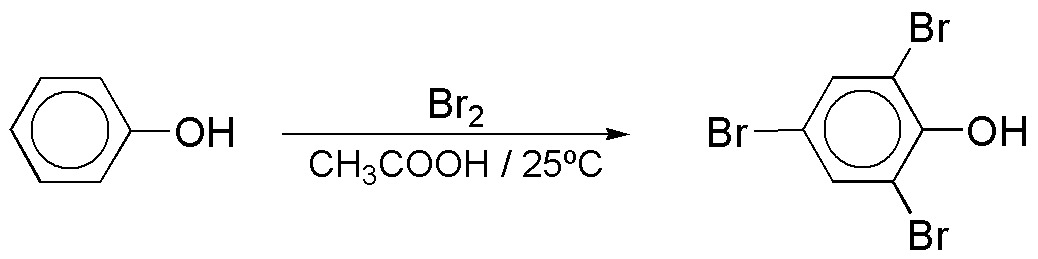
c) The bromination reaction of phenol leads to the formation of polysubstitution products, because the OH group is strongly activating.
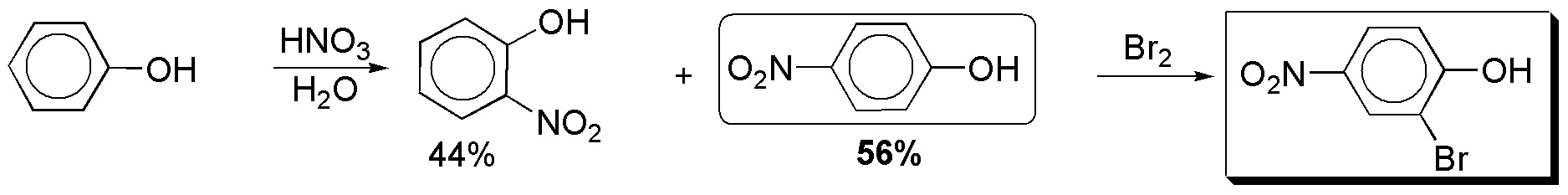
d) The nitration of phenol gives rise to a mixture of isomers, the proportion of the 1,4- compound being slightly higher than that of the 1,2- compound. From p-nitrophenol the desired product is obtained, since the 2-position of the benzene ring is especially activated.
Solution 7:
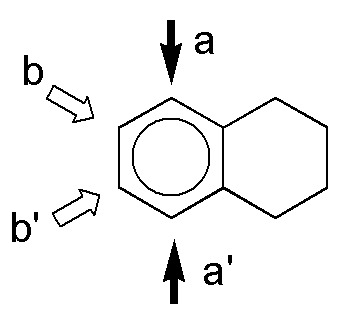
a) The positions that are activated are the ortho– and para– positions with respect to the substituents, marked with an arrow. The positions a and a’ and b and b’ are equivalent. The major product is obtained by substitution at b or b’, for steric reasons. As the two substituents are moderately activating, the reaction will be more favored than in the case of benzene.
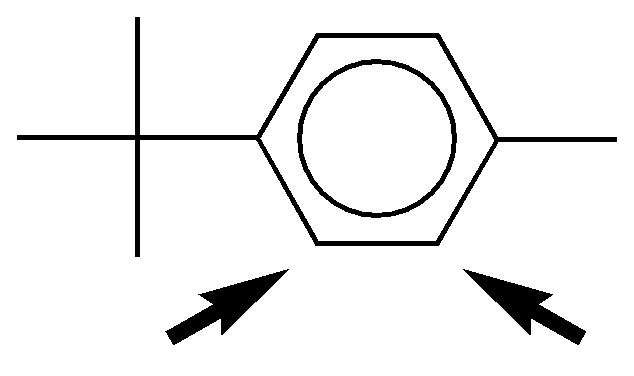
b) The methyl and tert-butyl groups are ortho– and para-directive. Due to steric effects the substitution in the ortho-position predominates with respect to methyl. The ring is richer in electrons than benzene so electrophilic substitution is favored.
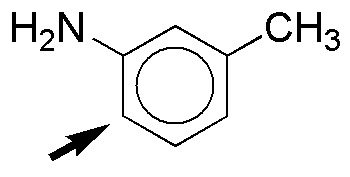
c) The amino group, NH2, is strongly activating while the methyl group, CH3, is weakly activating. One of the ortho-positions is doubly favored with respect to the amino group (marked with an arrow), since in this case there is a cooperative effect. The reaction rate will be higher than that of benzene.
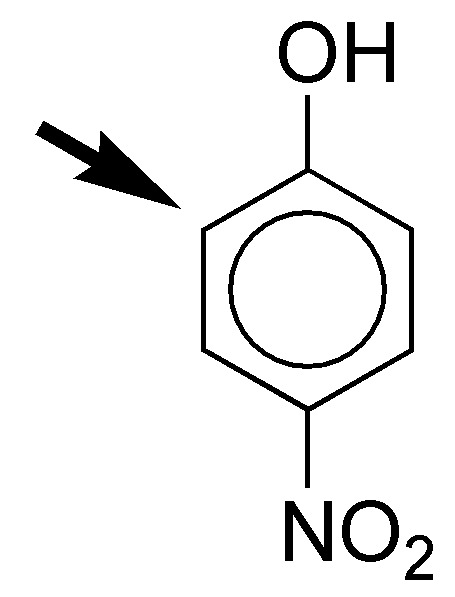
d) The two substituents have an activating effect on the aromatic ring. The reaction rate will be higher than in benzene itself. The major product of the substitution will be in the para-position with respect to the OH group, since the effect of this group prevails because it is strongly activating and the marked position is less sterically impeded.
e) The acetamido group is moderately activating and the carboxyl group moderately deactivating. The rate of electrophilic aromatic substitution will be similar to that of benzene. As for the orientation, the groups show a cooperative effect, since the position marked with the arrow is doubly activated by the para-directing effect of the acetamido and meta-directing effect of the COOH group.
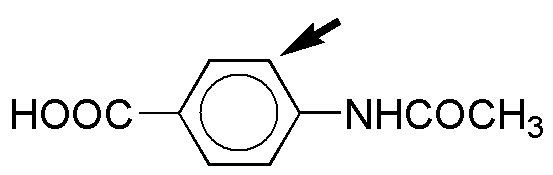
f) Both groups are activating, so that the substitution reaction will be much faster than in the case of benzene, however, a mixture of products will be obtained, since none of the groups is particularly conducive to attack in one of the free positions (antagonistic effect), in any case with a higher percentage of ortho-substitution product with respect to the NH2 group, since it is strongly activating, while the OCH3 group is moderately activating.
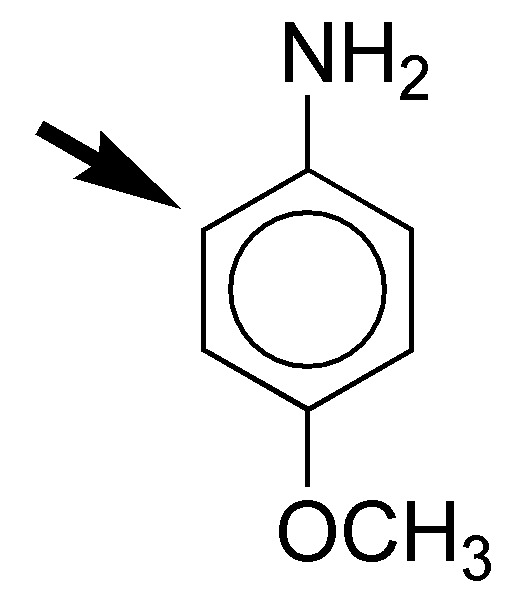
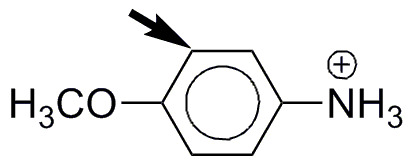
g) An ammonium salt is strongly deactivating, since it decreases the electron density of the aromatic ring. In contrast, the OCH3 group is moderately activating. The rate of substitution reaction should be somewhat lower than in the case of benzene. The ortho-position, with respect to the activating group the most favored in the substitution reaction.
Solution 8:
a) Hydrogenation with Pd(C) results in the addition of hydrogen on the double bond of the chain, while the benzene ring remains unchanged. In order to hydrogenate an aromatic ring, high hydrogen pressures and high temperatures are required, thus the result of the reaction is as described.
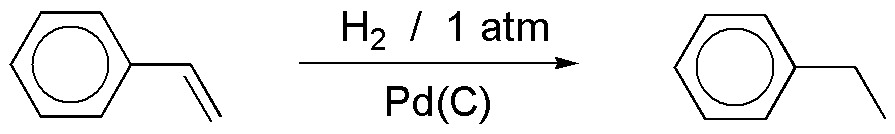
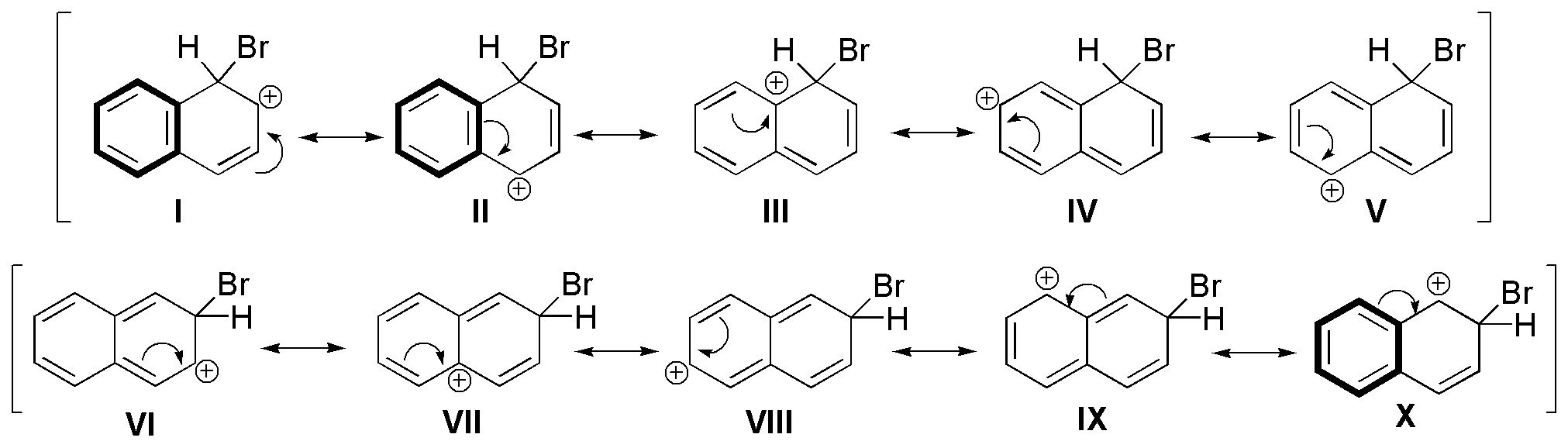
b) The halogenation of naphthalene can give, in principle, two types of products, with substitution at C1 and at C2.

The product obtained is mostly 1-bromonaphthalene. The higher reactivity of position 1 is due to the difference between the two reaction intermediates postulated for each of the attacks.
In both cases we can find five resonant structures, but with different energies, since for the attack in position C1, two structures that retain the aromaticity of the second ring are maintained (marked in thick stroke), while for the attack in position C2, only one. Since the aromaticity-preserving structures contribute more to the hybrid by resonance, the attack at C1 is more favored than at C2.
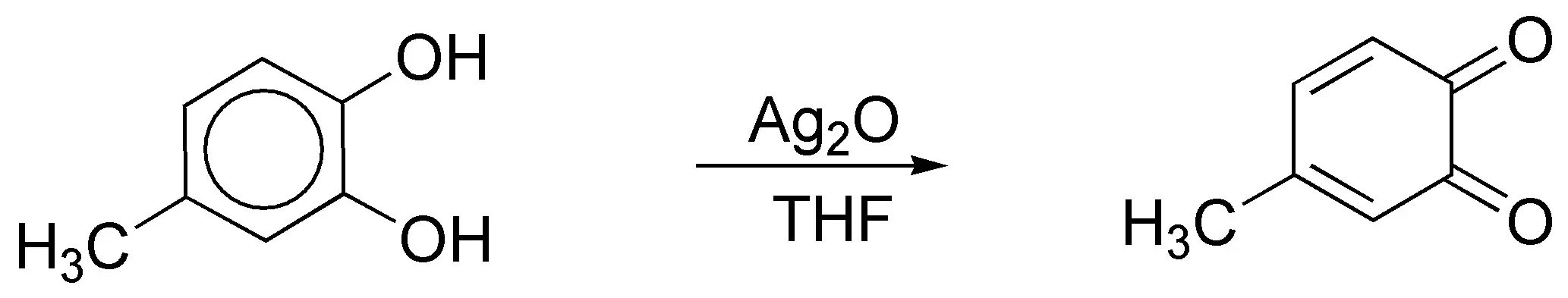
c) Treatment of a 1,2-diphenol (pyrocatechol) and its derivatives with silver oxide in ether or THF leads to the formation of 1,2-quinones, so the product obtained will be:
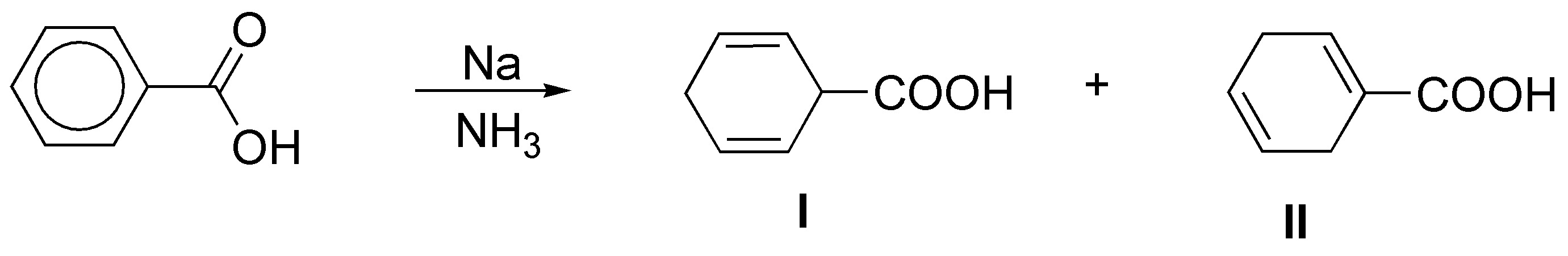
d) Sodium in ammonia is a reducing agent that transforms benzene rings into cyclohexa-1,4-dienes. This procedure is compatible with substituents on the ring such as -COOH, -COOR, -CN, alkyl groups, -OR or -NH2 among others.
In monosubstituted benzenes, such as the one in the problem, there are two possibilities regarding the formation of the final product (I and II).
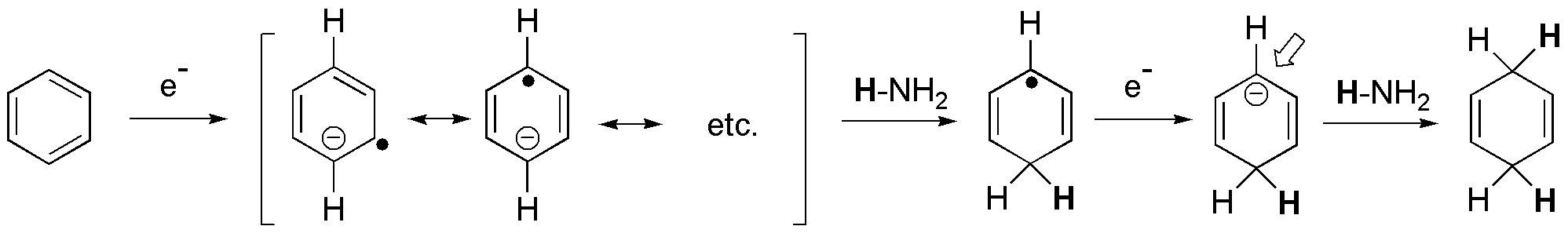
The product obtained can be predicted, considering the mechanism that justifies the reaction being regioselective, since the substituents with electron-attracting effect (-M effect) stabilize the charge in the position marked with an arrow, while the electron-donating groups (+I effect) destabilize it. This fact conditions the positions in which the double bonds remain.
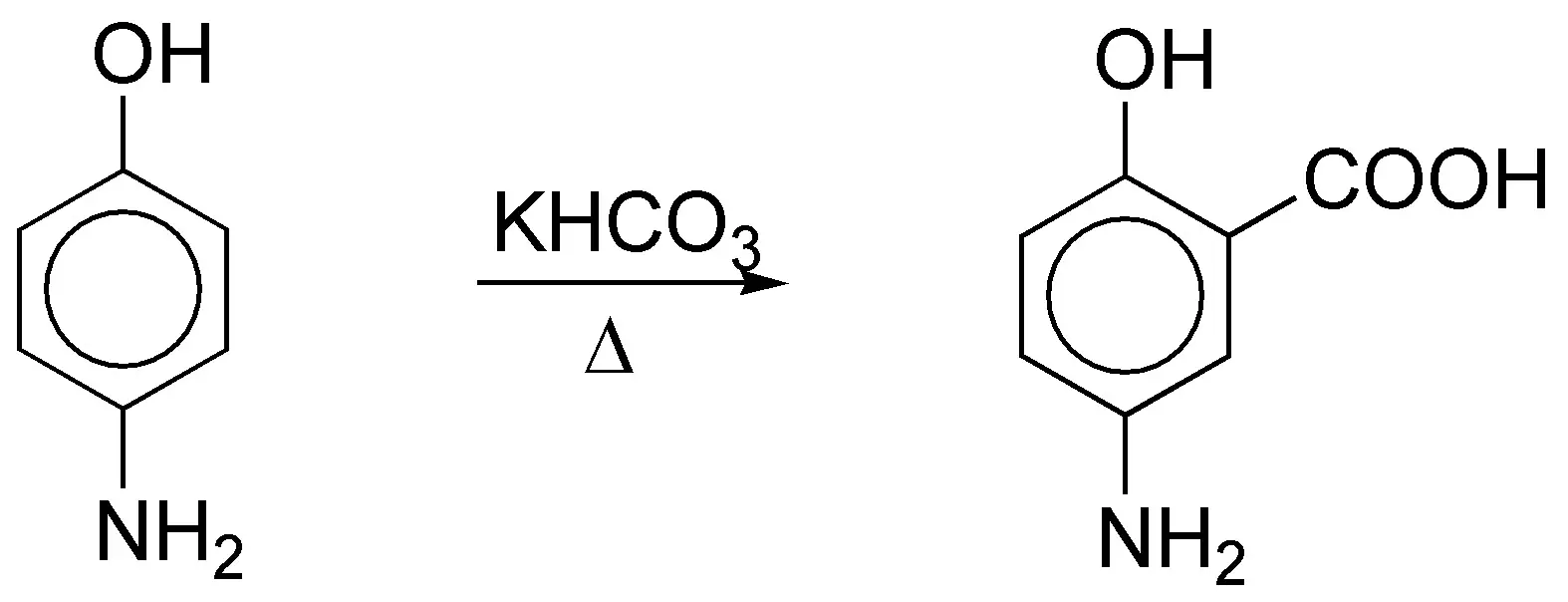
e) The bicarbonate ion decomposes thermally to give CO32- and CO2 according to the equation:
![]()
![]()
Phenols give salicylic acid derivatives by Kolbe-Schmitt reaction. In this case the product obtained will be:
f) In cases like this the two rings must be considered separately. Ring A is less reactive than ring B in an electrophilic aromatic substitution reaction, since it has a moderately deactivating substituent and ring B has the same group and another moderately activating group such as isopropyl, which is the one that conditions the substitution. The two most reactive positions are the ortho– and para– with respect to this group. Since the para-position is occupied, the bromination product will be the ortho– shown:
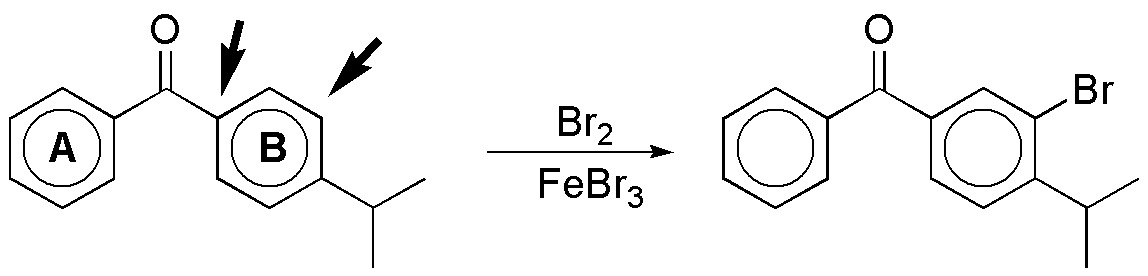
Solution 9:
In the nucleophilic aromatic substitution mechanism by an addition-elimination process, there is an increase in the electron density at the ortho– and para– positions with respect to the group being substituted. The greater the number of clusters capable of stabilizing the charge.
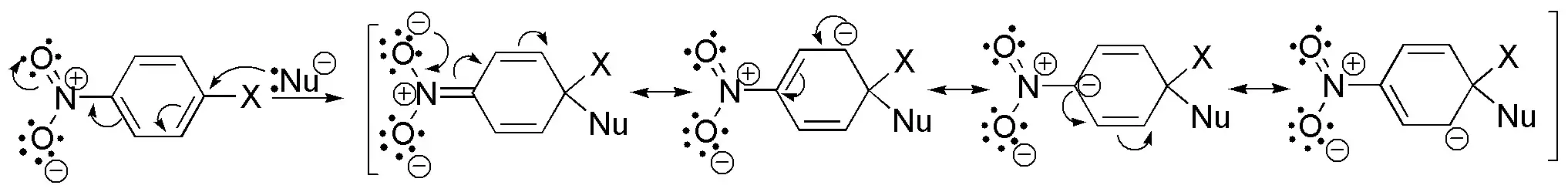
On the other hand, the reaction rate is a function of the leaving group according to the order F > Cl > Br > I. Taking these factors into account the order will be:
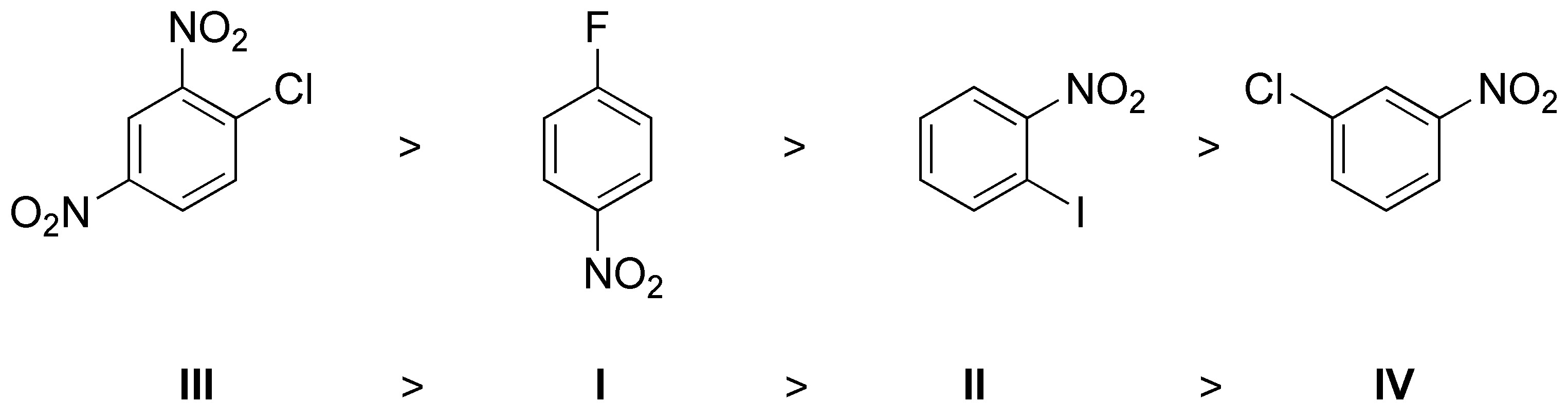
Solution 10:
a) The final product will be the result of a nucleophilic aromatic substitution reaction via benzene, i.e., via an elimination-addition mechanism. The amidide, acts as a base and nucleophile. Since we start from a substrate with a substituent, we obtain a mixture of products.
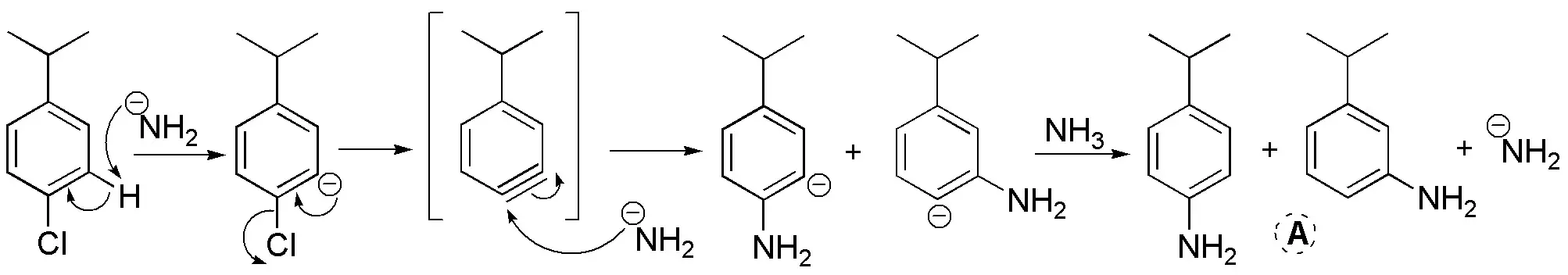
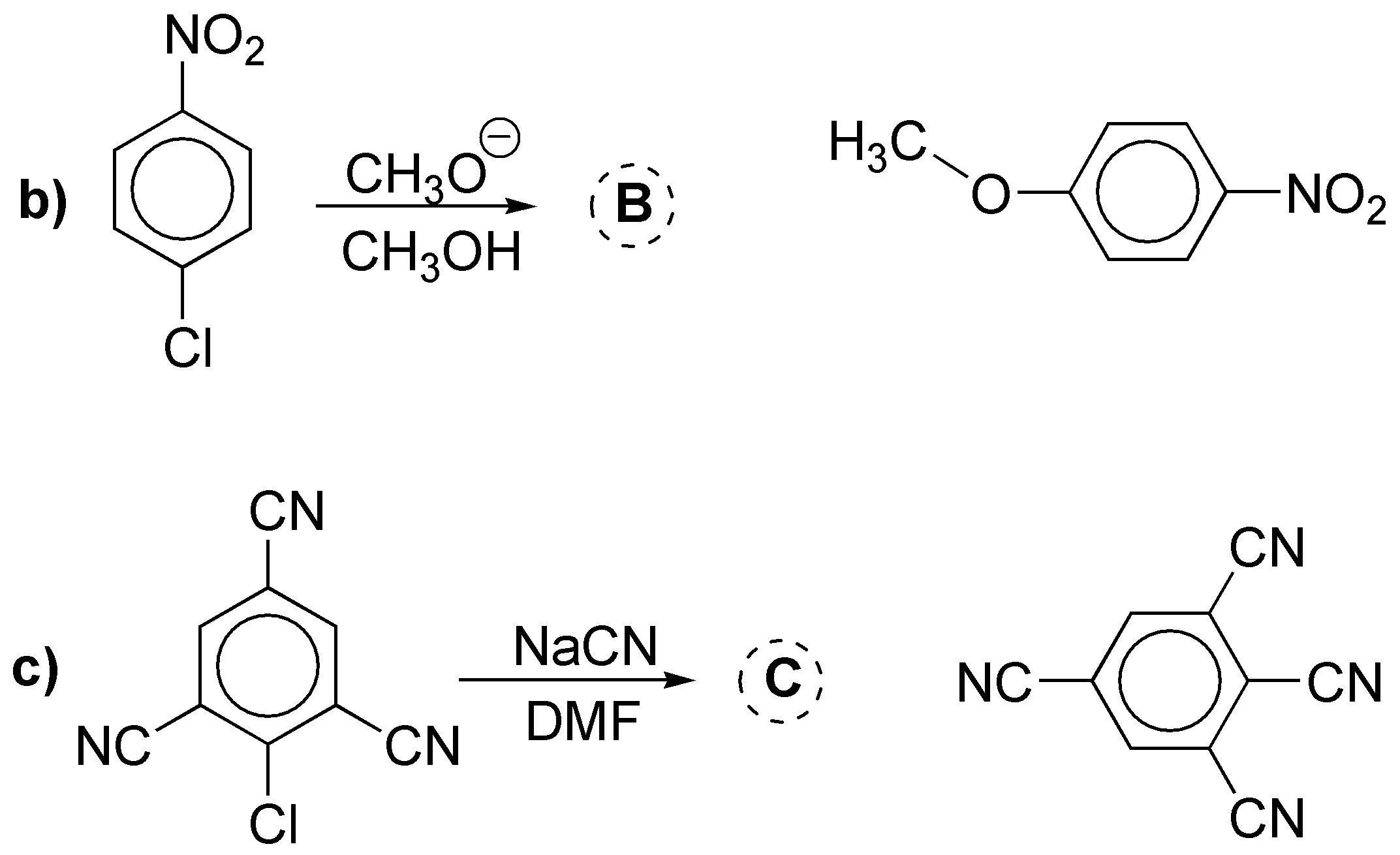
b) and c) are nucleophilic aromatic substitution reactions with addition-elimination type mechanisms. Both substrates are compatible with this kind of reactions, since they present electron-attracting groups in the ortho– and/or para– positions with respect to the leaving group. The products obtained will be:
Solution 11:
The rate of the reaction will depend on how activated the benzene ring is. In view of the groups present the reactivity will be:
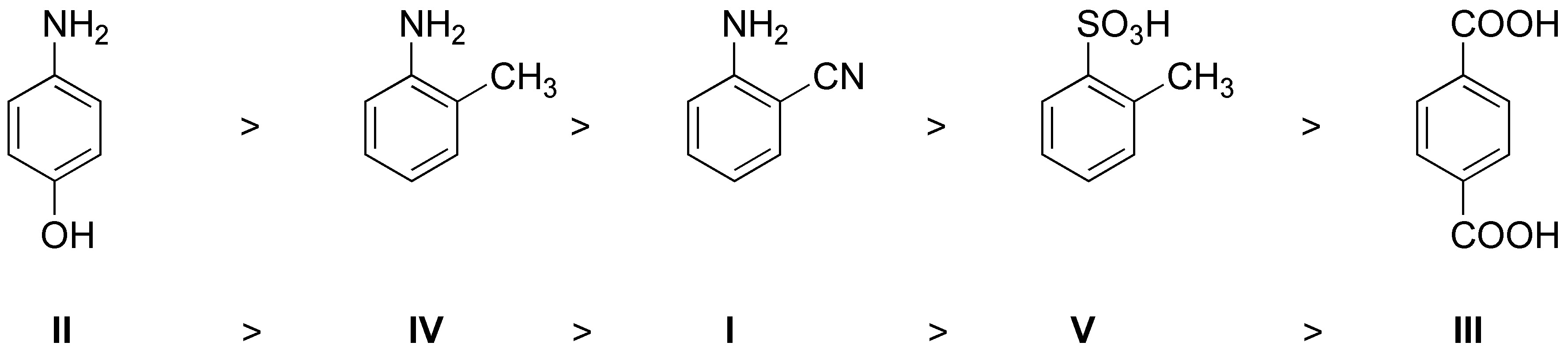
Solution 12:
Those groups that are electron-attracting or possess a positive charge (net or partial) on the atom attached to the aromatic ring (the exception is the NO (nitrous) group) in this case will be the groups: b) CH2Cl, c) COOH, d) SO3H and e) CONH2; the others will be ortho– and para-oriented.
Solution 13:
Bromination of aniline and acetanilide give 2,4,6-tribromoaniline and 4-bromoacetanilide, respectively.
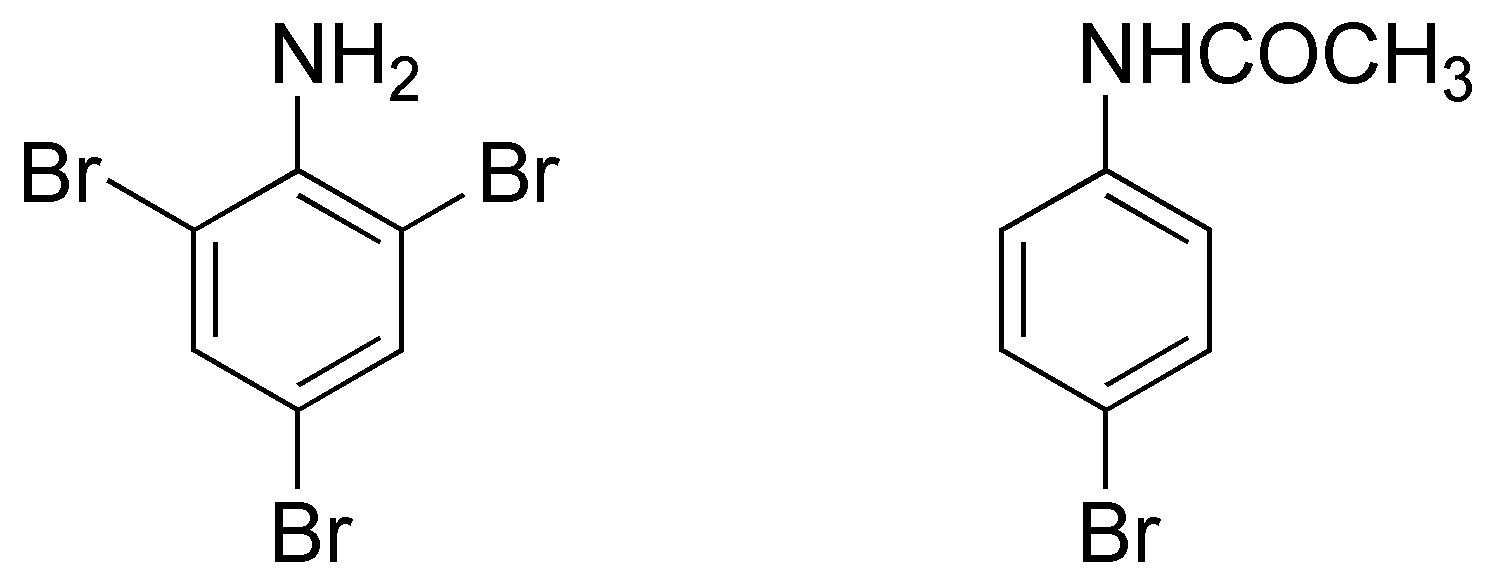
The reason for this behavior is that the amino group is such a strong activator that the reaction does not stop at the monobrominated derivative, but produces bromination at all activated positions. In contrast, the acetanilide has a milder activator and produces, mostly, the monoderivative in para due to steric hindrance.
Solution 14:
The arrows in b) indicate two identical positions.
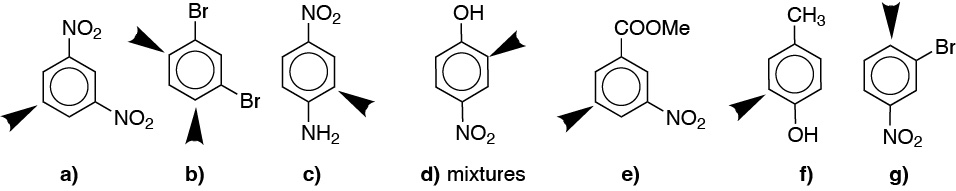
Solution 15:
Two opposite factors are given for the nitro (NO2) and nitroso (NO) group. On the one hand, the intermediate carbocation formed in the addition step is destabilized in the case of the nitro group in the ortho– and para-positions, because it has two adjacent positive charges. On the other hand, the attack at ortho– and para– is especially stabilized in the case of the nitro group, since the nitrogen can yield a pair of electrons to the adjacent carbon, to form another stabilized resonant structure. This does not occur with the nitrous group in meta– since the carbon adjacent to the nitrogen is not positively charged. The sum of the two effects causes the nitro group to lead the attack at meta– while the nitro group leads the attack at ortho– and para-.
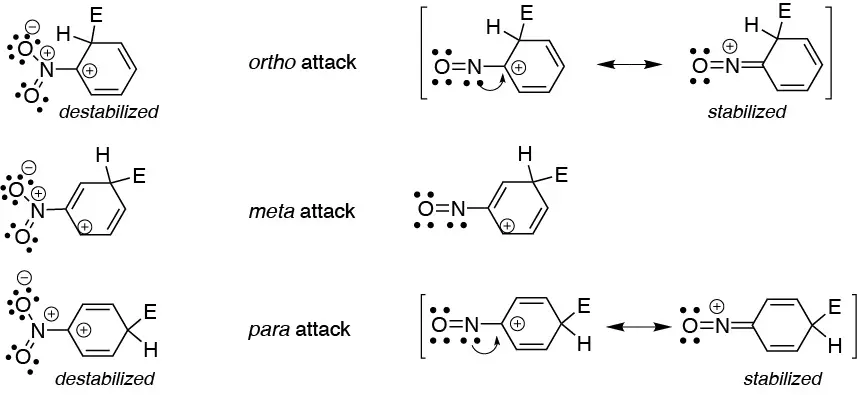
Solution 16:
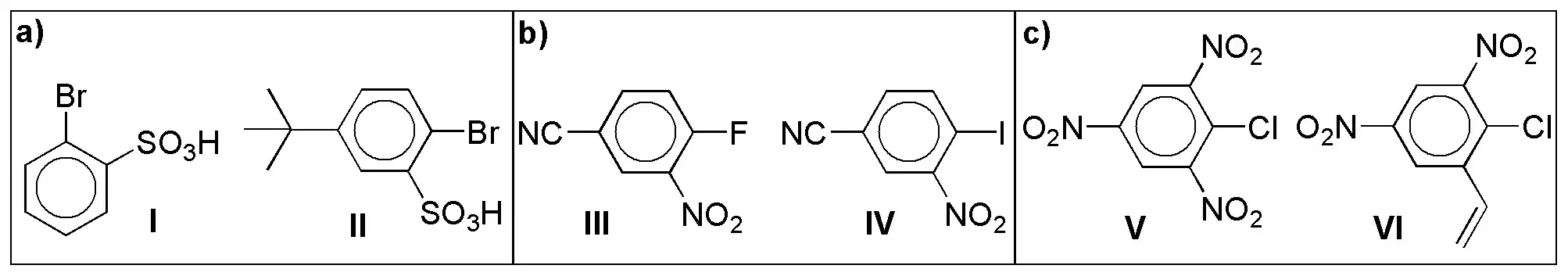 The less deactivated ring, i.e. the one with the least deactivating groups, will react faster, therefore the solution will be:
The less deactivated ring, i.e. the one with the least deactivating groups, will react faster, therefore the solution will be:
I > II
IV > III
VI > V
Solution 17:
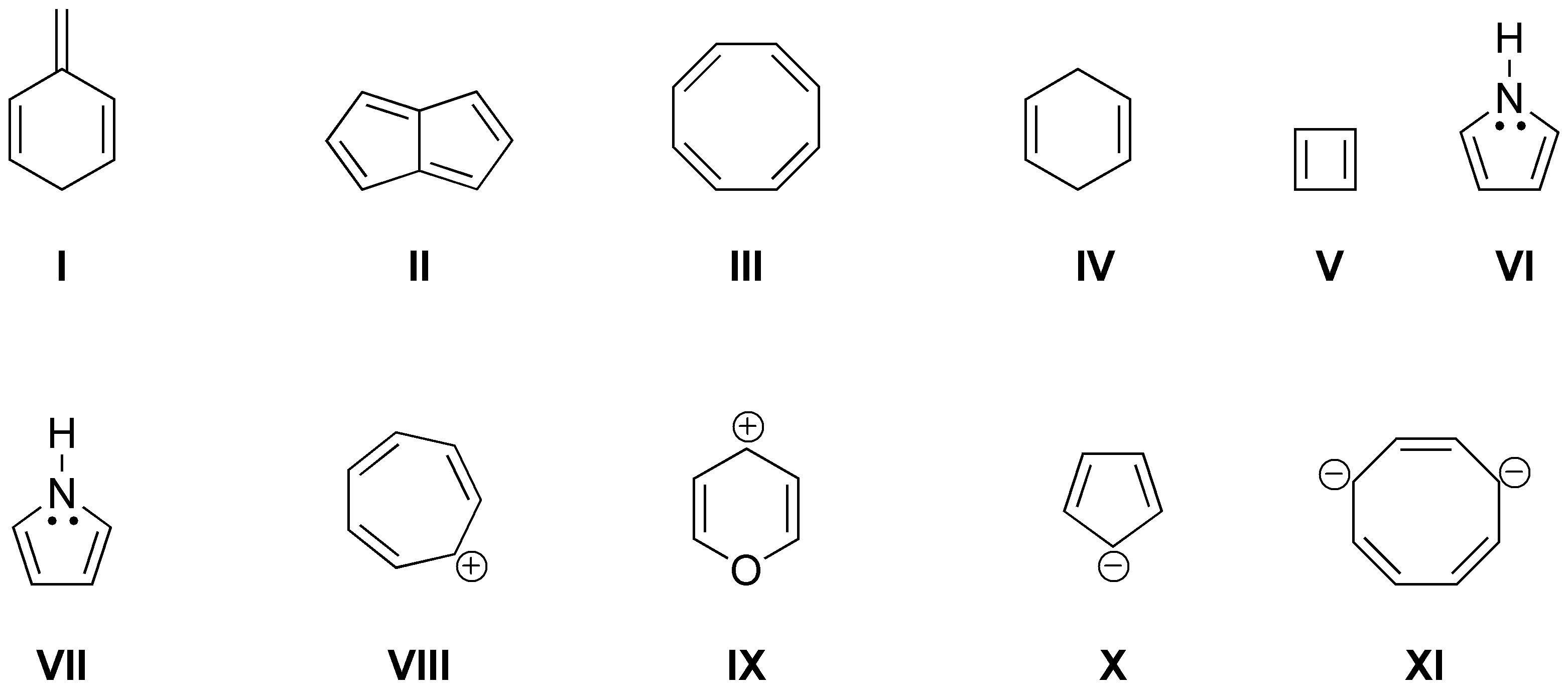
Will be non-aromatic products I, III and IV, for not being conjugated or not being flat, antiaromatic products II, V, VII, for not complying with Hückel’s rule and aromatic the remaining ones: VI, VIII, IX, X and XI, because they do fulfill it.
Solution 18:
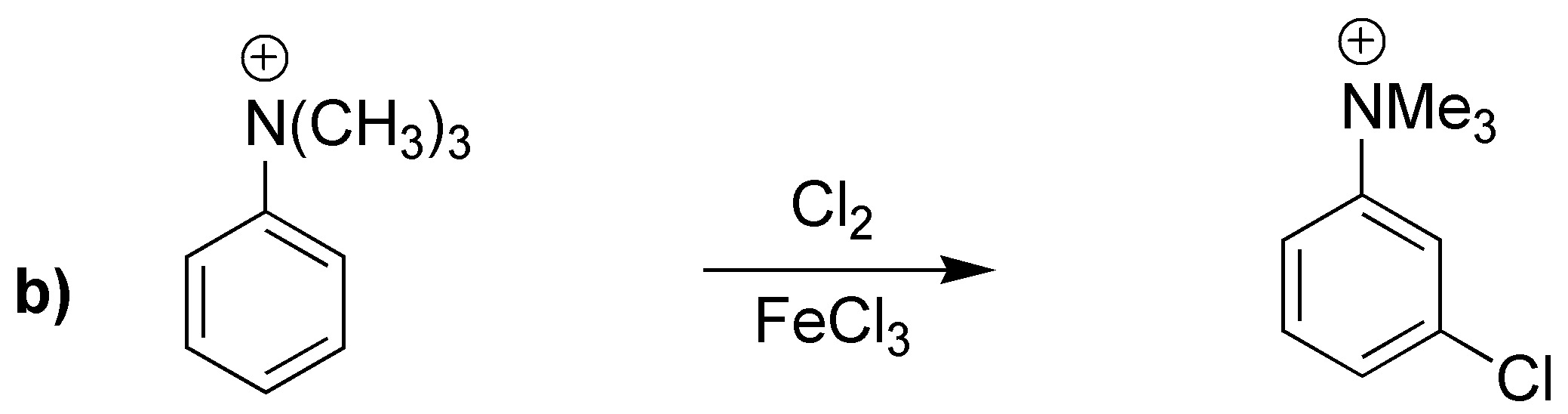
Reactions a) and b) lead to substitution products in the meta-position because they possess electron-attracting (deactivating) groups. The product obtained in c) has an activating group (cyclohexyl) and by steric hindrance will produce mostly the para– derivative.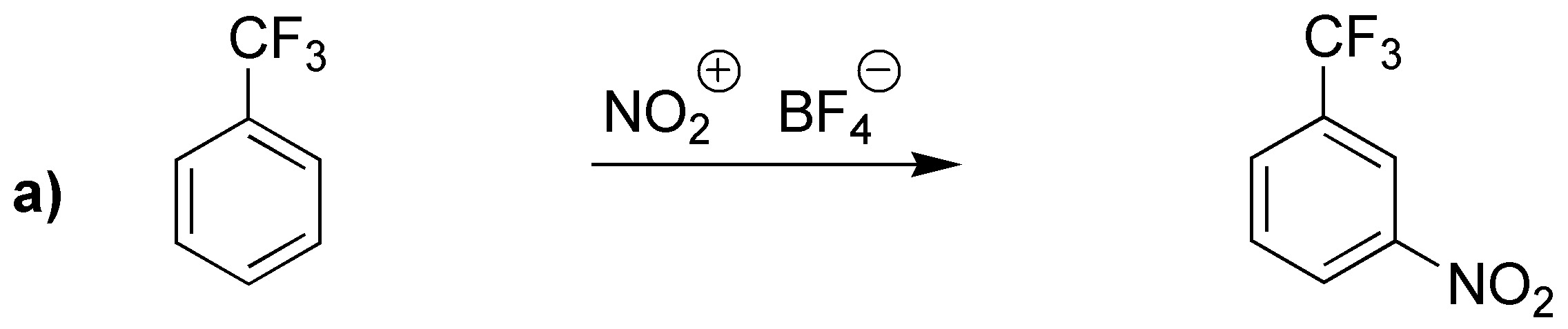
![]()
![]()
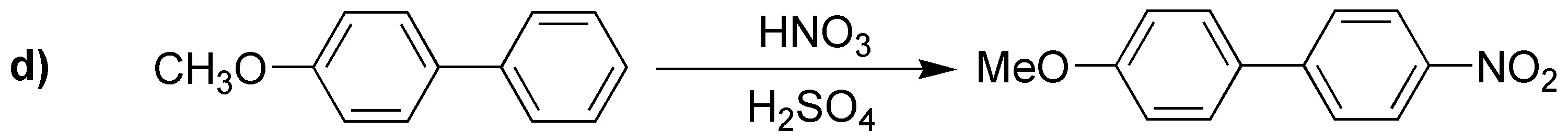
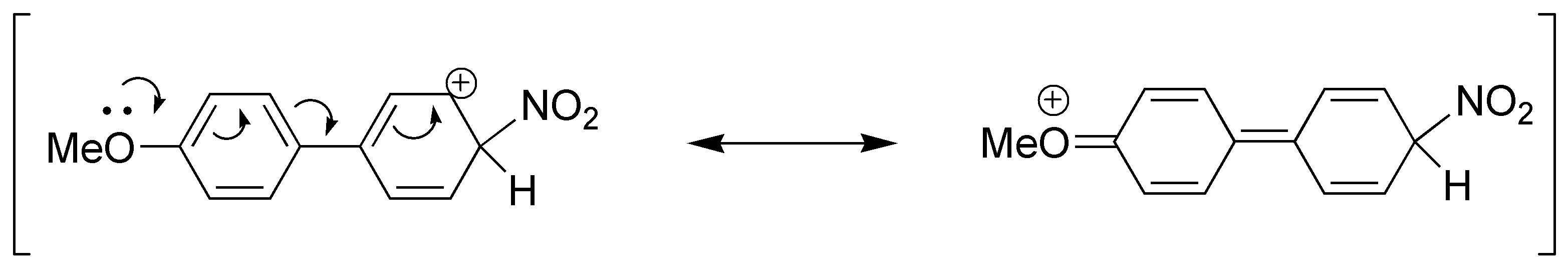
The product obtained in d) will be the indicated one because it possesses the intermediate shown which is very stable.
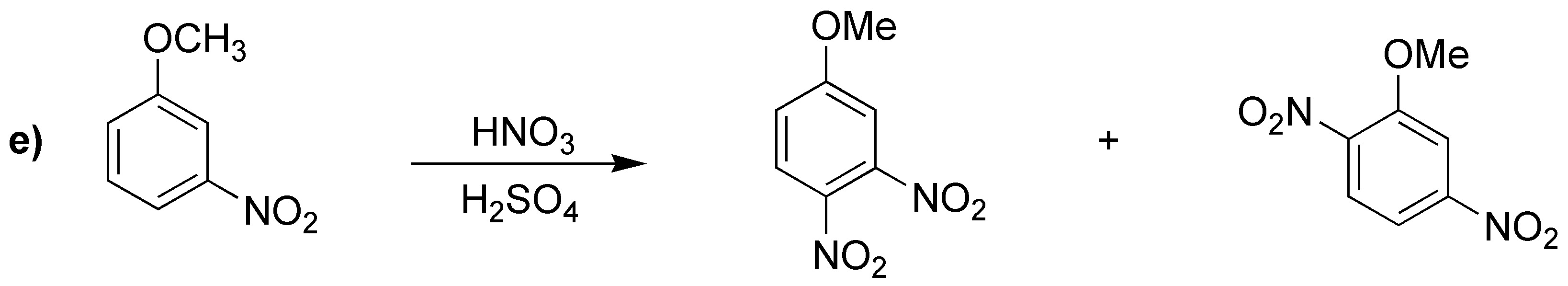
In reaction e), a mixture of products will be produced taking into account that it will be the methoxyl group that controls the orientation as it is activating.
Solution 19:
The orientation of the substitution will be determined by the character of the substituents. The most activating one determines the position of the substitution.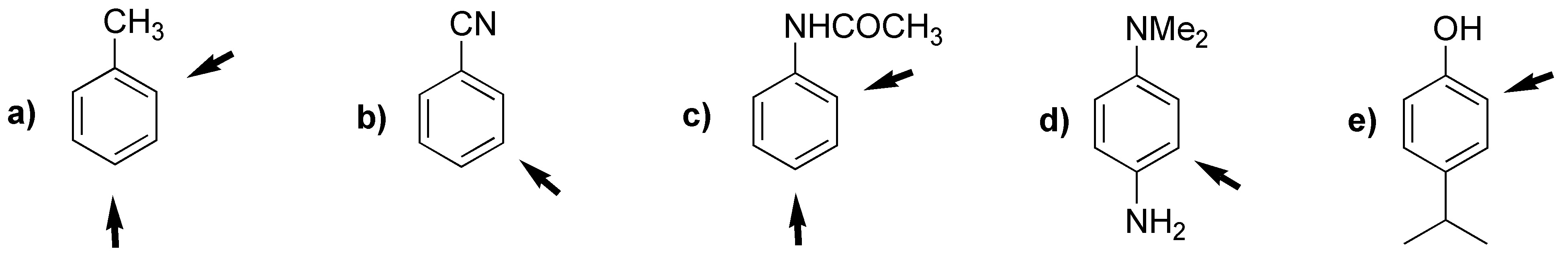
Solution 20:
Reactions a), b) and c) correspond to three Friedel-Craft alkylations. while d) is an acylation.
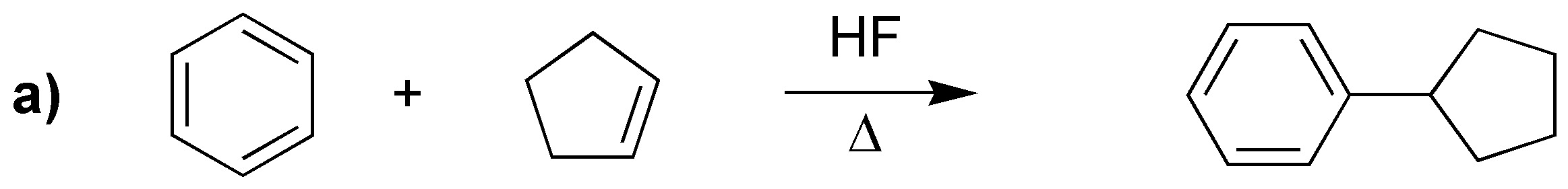
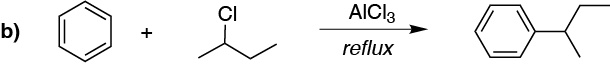
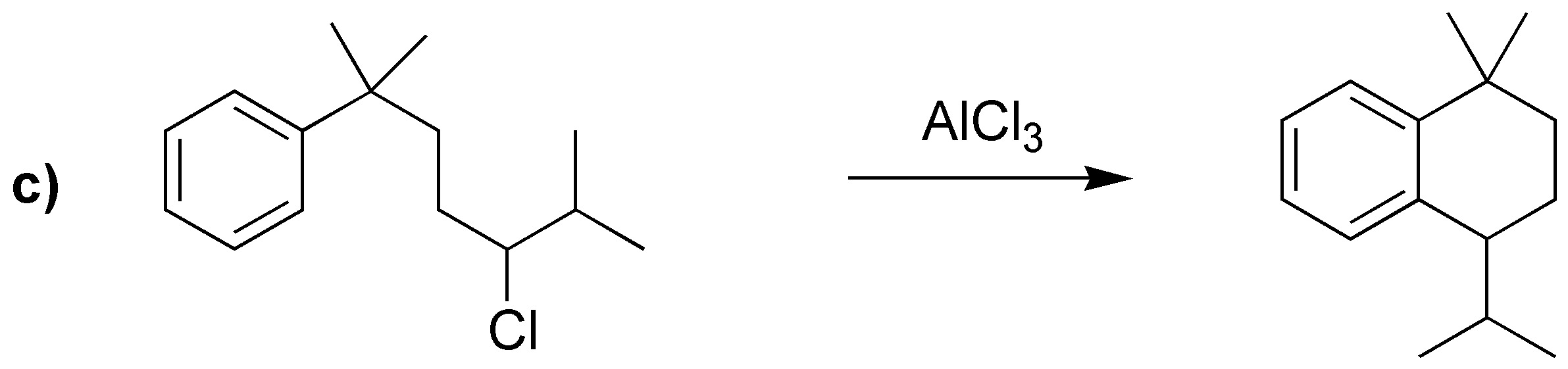
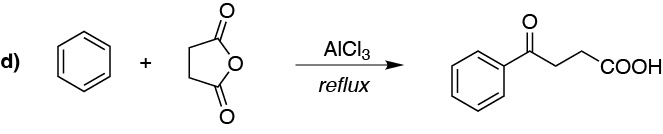
Solution 21:
Producing only one derivative, product A must be para-xylene. Product B yielding two derivatives will be ortho-xylene. While C will be meta-xylene, as shown in the figure:
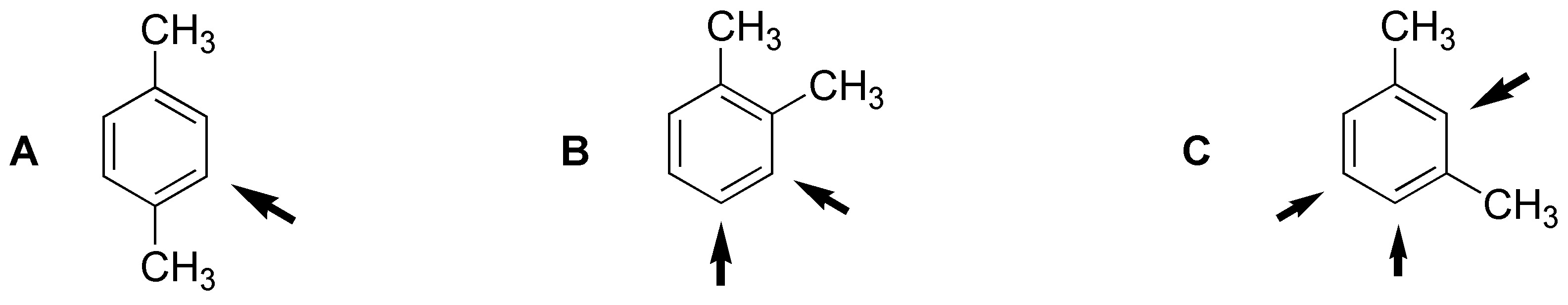
Solution 22:
Toluene will react faster than benzene as it possesses a methyl group which is an activator (like all alkyl groups), this translates into higher speed, lower temperature needed for the reaction to occur or milder reaction conditions.
Solution 23:
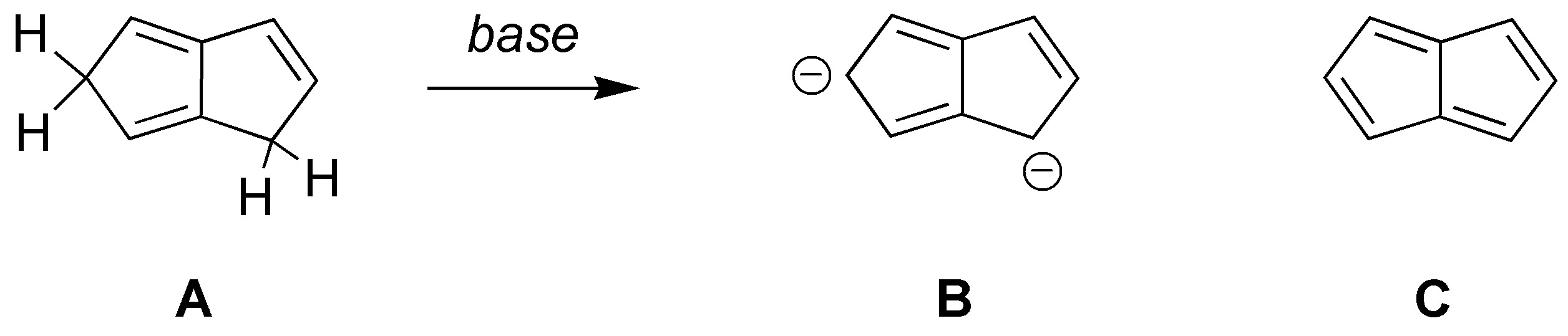
The dianion B produced in the reaction is stable as it is aromatic by fulfilling Hückel’s rule. Tetraene C on the other hand does not fulfill it, it is antiaromatic and therefore is not stable.
Solution 24:
Birch reduction will yield the indicated dienes:
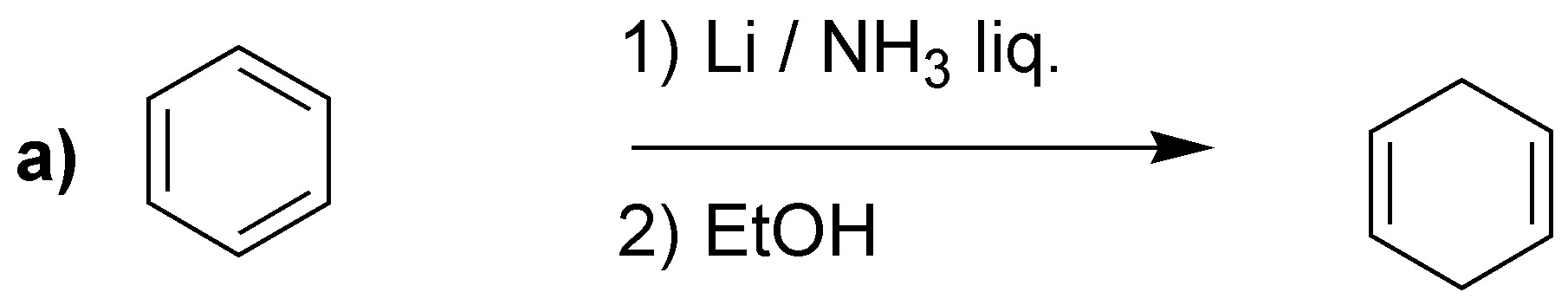
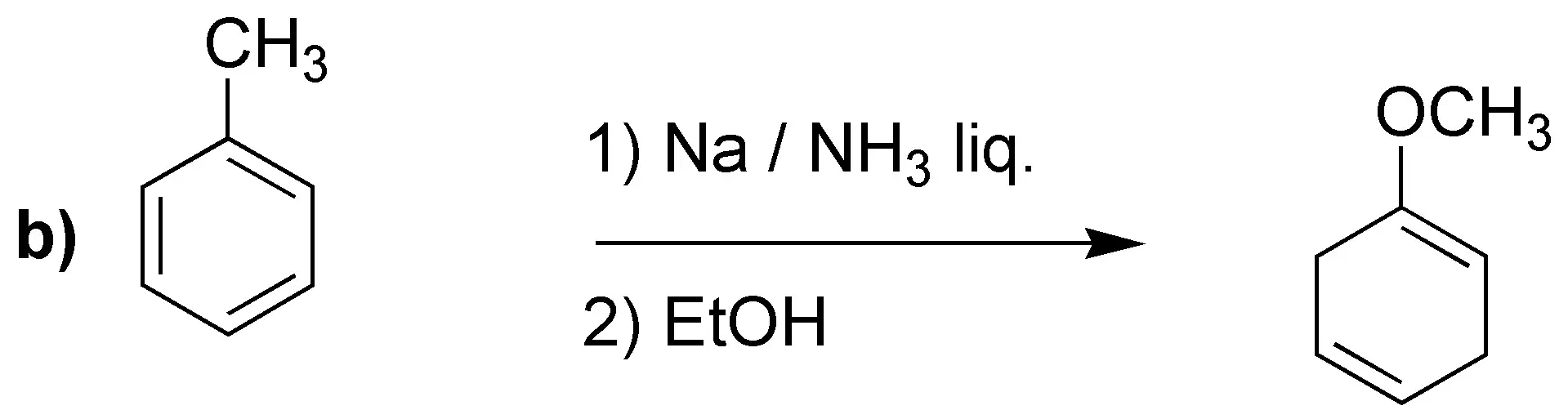
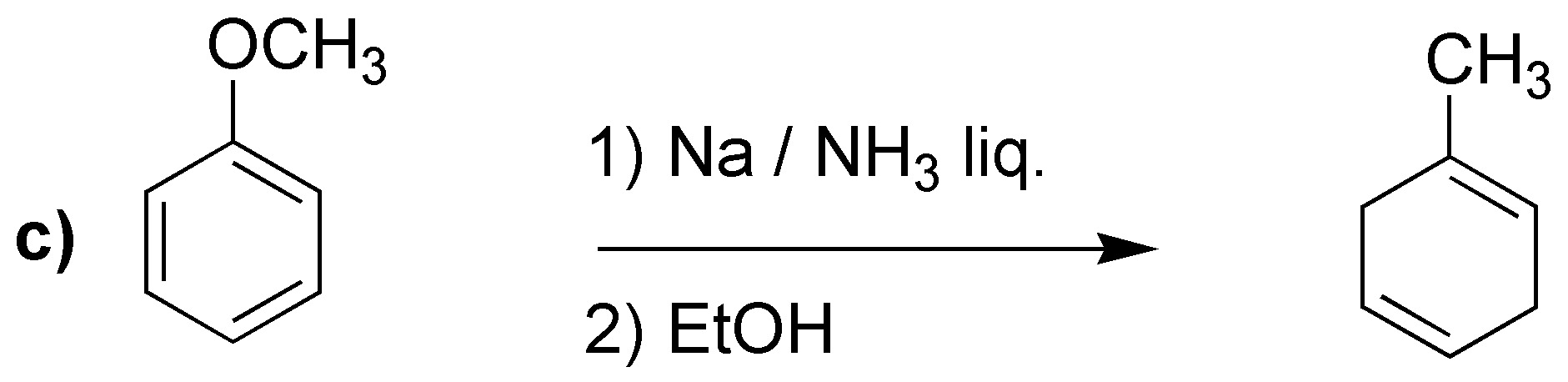
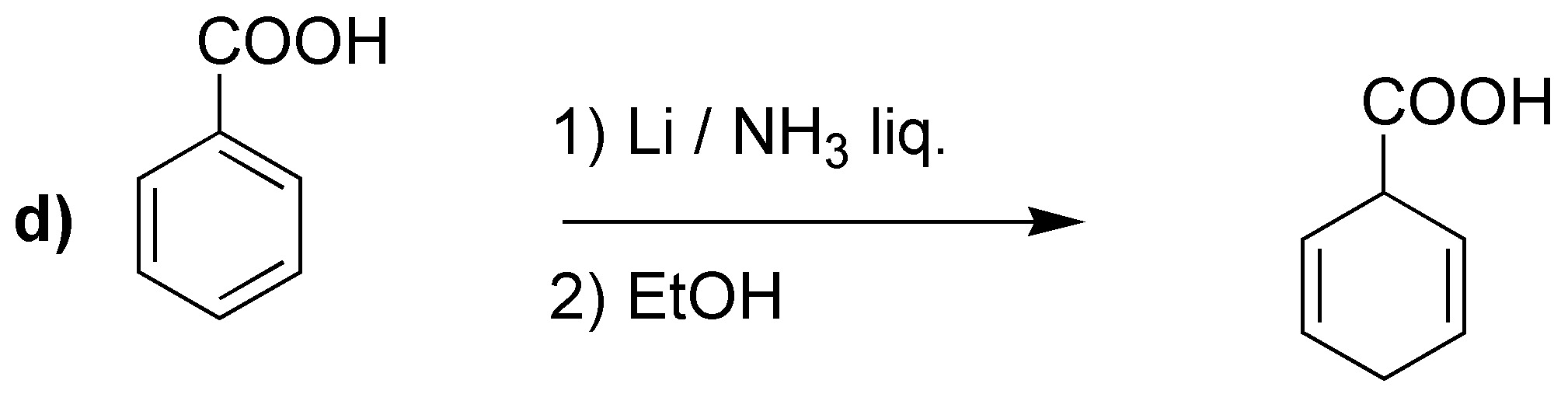
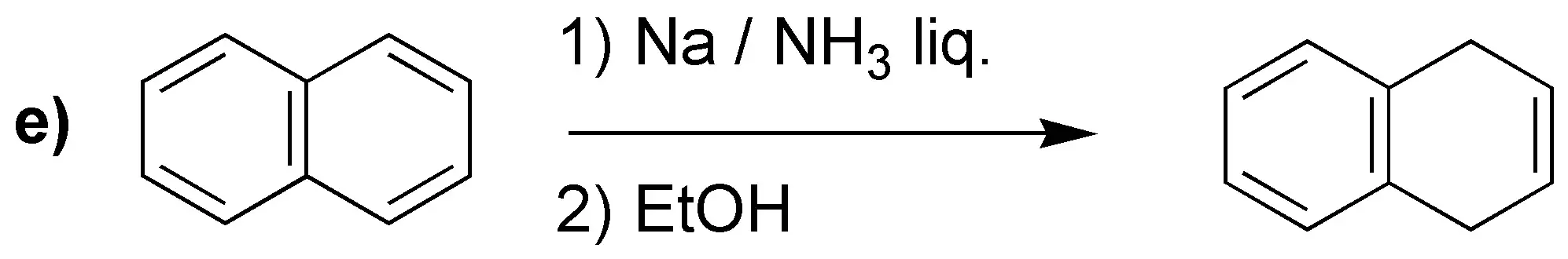
It is worth drawing attention to case d), where unlike the previous cases, the most substituted double bond is not obtained. It has been proposed as an explanation that electron attracting groups would be responsible, as in this case -COOH.
Solution 25:
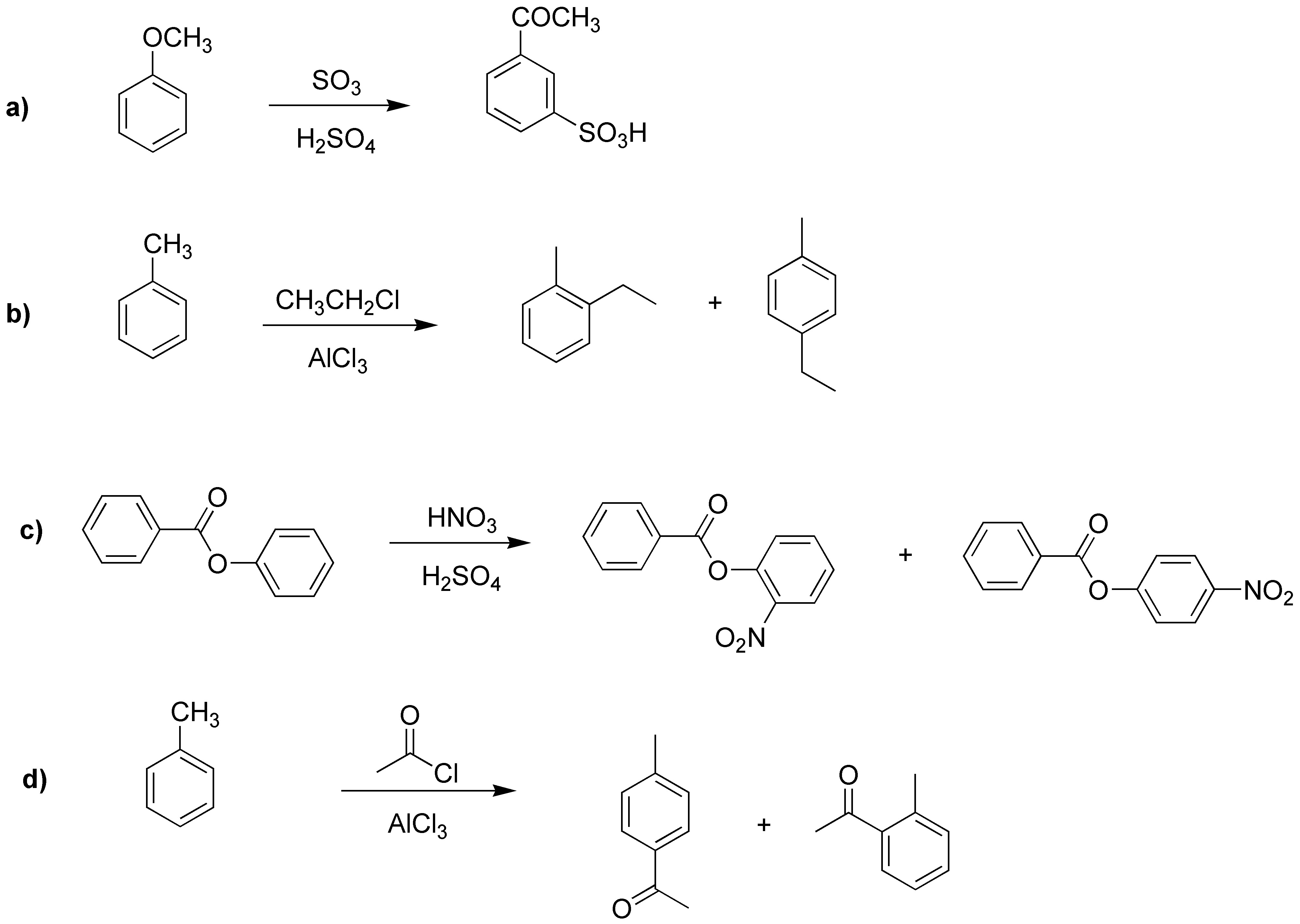
Process a) corresponds to a sulfonation and gives as product a sulfonic acid; b) is an alkylation of toluene which will produce the mixture of the two possible isomers. In case c) the reaction will occur on the second benzene ring because it is activated while the first is deactivated, resulting in a mixture of nitro compounds. In case d), the acylation will produce a mixture of compounds where the major one will be the para-product.
Solution 26:
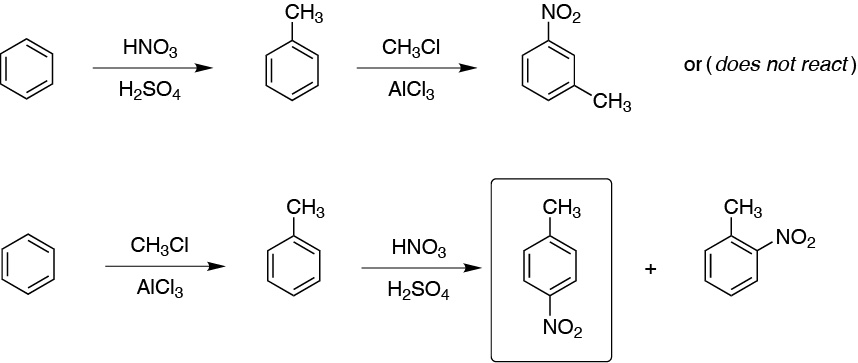
Since the final product has a para arrangement of substituents it will be necessary to first introduce the methyl group which is ortho and para leading. Subsequent nitration of the same will produce a mixture of both isomers in which the para will be in the majority due to steric hindrance. If the reactions were reversed, the product produced would be meta-nitrotoluene, if such a process could occur.
Full Professor of Organic Chemistry at the University of Granada, with a long-standing research career in Computational Chemistry and molecular modeling and design.