
Introduction
The structural determination of an organic substance will always start with the purchase, synthesis or isolation of “a pure product” and the following aspects should be considered:
Separation, isolation and purification
We should therefore begin (in the most general approach) by recalling the physical methods of “separation, isolation and purification” of organic compounds:
- Crystallization.
- Distillation in its different modalities: simple, fractionated, vacuum, steam distillation.
- Sublimation.
- Extraction.
- Chromatography, also in its various forms: partition, absorption, gas, exclusion or ion exchange chromatography.
- Electrophoresis, etc..
Physical characterization
Once the organic product has been isolated (or purified) we should characterize it both by its physical properties and by chemical methods, so in the physical characterization, we should/could determine:
- Organoleptic properties: odor, color, flavor
- Melting point (if the product is solid)
- Freezing point
- Boiling point if liquid
- Density
- Molecular weight
- Refractive index
- Optical rotation (if the product is of natural origin), etc.
Chemical characterization
In the chemical characterization, we could consider:
- Qualitative and quantitative elemental analysis.
- Classification by solubility.
- Determination of acidity or basicity.
- Identification of functional groups.
- Preparation of derivatives.
Once this chemical-physical characterization has been carried out, we should compare the data obtained with the data collected in the bibliography (data tables or specialized bibliography).
The main problem of this determination routine is the time needed to perform it, which is normally very long, and that is why the structural determination is currently performed by means of spectroscopic techniques, many of which even allow us to avoid the tedious step of purification and isolation of the product, being its main limitation the high cost of the instruments and their maintenance, as well as the need of specialized personnel to interpret the data obtained.
Considering the chronological development of the different spectroscopic techniques, their first function was to try to shorten the time of the structural determination, later they became the data base for the characterization of organic products and finally they have become the fastest and most precise instrument for the structural determination that the organic chemist has.
It is from this last point of view that this tutorial will be oriented.
Basic concepts
The following is a brief review of a series of definitions, concepts and formulas that we consider useful when identifying an organic compound from its spectroscopic data (IR, MS, 13C-RMN y 1H-RMN).
For brevity, UV/vis spectroscopy has been intentionally omitted because we consider that it provides very little structural information compared to, for example, NMR or IR.
To learn more about each of the techniques, the reader is encouraged to consult some of the many books or monographs available on the subject.
The electromagnetic spectrum
All radiation is characterized by a wavelength (λ), a frequency (ν) or an energy (E), being the relationship between them:
E = h·ν = h·c/λ
The first question to ask ourselves is what happens to matter when it is subjected to a certain radiation. As we can imagine, this effect will depend on the energy of the radiation. Table 1 shows the immediate effects on matter, from the highest to the lowest energy:
| Radiation | Effect |
| X-rays and cosmic rays | Ionizations of molecules |
| UV-Visible | Electronic transitions between atomic and molecular orbitals |
| Infrared (IR) | Deformation of chemical bonds |
| Microwaves | Rotations of chemical bonds |
| Radio frequencies | Electronic or nuclear spin transitions in the atoms of the molecule. |
When radiation is incident on a substance, not all of it is affected by it. For example, the atom or set of atoms that absorb radiation is called a chromophore and in each spectroscopic technique it will be different within the same molecule. Auxochromes are a type of chromophores that do not absorb radiation, but cause some of the absorption characteristics to be modified.
Spectroscopic techniques
The second question is how we can use such effects on substances to obtain information about the structure of matter and how to use this information.
In a very schematic way, the most important characteristics of the spectroscopic methods are the following:
| Spectroscopic technique | Information obtained |
| X-Ray | Total structure of the molecule including the stereochemistry of the molecule from the relative positions of the atoms. |
| UV/vis | Existence of chromophores and/or conjugation in the molecule from the observed absorptions |
| IR | Functional groups from observed absorptions |
| (MS)to | Molecular formula and substructures from the observed ions |
| NMR | Functional groups, substructures, connectivities, stereochemistry, etc., from chemical shift data, peak areas and observed coupling constants |
| aMass spectrometry is not a spectroscopic technique in the sense that we are seeing because there is no electromagnetic irradiation of the substance and there is no absorption of such radiation. | |
The question of which technique to use or in what order, if several, to achieve the complete structural determination of a substance will depend largely on the objectives pursued.
In general terms, we can indicate the following as successive objectives:
- Molecular formula.
- Identity of functional groups.
- Carbons connectivities.
- Positioning of substituents and/or functional groups on the carbon skeleton (obtaining substructures).
- Stereochemical properties including both static and dynamic aspects.
Structural determination objectives
Expressed in another way, we can indicate as objectives of the structural determination the elucidation of the Stereochemistry of the molecule, understanding as such the spatial arrangement of the atoms that form it, and that implies the successive knowledge of:
- Composition: atoms present and their proportion in the molecule, which translates into a molecular formula.
- Constitution: Existing bonds between atoms, which translates into the determination of the functional groups and substructures present in the same.
- Configuration: spatial arrangement of atoms in the molecule. y
- Conformation: spatial arrangement of the molecule that arises due to the possibility of rotation or rotation of the single bonds in the molecule.
Before addressing the resolution of a specific structural determination from the spectroscopic data of the same, we recommend that you go to the sections on the main characteristics of each of the spectroscopic techniques.
Degree of unsaturation (or IHD, Index of Hydrogen Deficiency)
IR spectroscopy
The IR spectrum is usually divided into four regions for better interpretation. Although some exceptions occur, such as the N-H bond deformation at 1550-1620 cm-1.
Region 4000-2500 cm-1
H-bonded single bond absorptions (C-H, O-H, N-H) occur.
In this region, free O-H deformations appear at 3600 cm-1 and for hydrogen bonds they appear in the range 3100-3200 cm-1.
Region 2500-2000 cm-1
The absorptions of triple bonds (C≡C and C≡N) and cumulative double bonds are given.
| Group | Absorption (cm-1) |
| -C≡C- | 2100-2300 |
| -C≡N | ≈ 2250 |
| -N=C=O | ≈ 2270 |
| -N=C=S | ≈ 2150 |
| >C=C=C< | ≈ 1950 |
Region 2000-1500 cm-1
Double bonds (C=C, C=O, C=N) generally absorb in this region. For example, hexene exhibits a sharp absorption peak due to the C=C vibration at 1640 cm-1.
Other groups absorbing in this region are C=N and C=O. Table-A7.1 of characteristic frequency ranges of the carbonyl (C=O) is attached in the Appendix, which can be used to identify the functional group to which it belongs, e.g. ether, ester, carboxylic acid, ketone, etc.
Region 1500-400 cm-1
This region is characterized by the absorption of other bond deformations (rotations, angles, etc.).
Absorptions due to deformations such as rotations, scissors and bonding depend on the combination of bonds in the molecule. This part of the spectrum is unique and characteristic for each compound and is often referred to as the fingerprint region.
It is rarely used to identify particular functional groups, but some generalizations can be made:
- The C-O bond absorbs at 1300-1020 cm-1 although the absorption here is not exclusive to the C-O bond.
- On the other hand, this fingerprint region can be used to identify particular molecules, since no other compound will have the same region (with the same absorption pattern). There are databases of spectra that include this region.
1H and 13C NMR spectroscopy
Chemical shift (δ)
It is the place in the spectrum where a certain signal appears. In addition, symmetry causes some hydrogens to appear at the same chemical shift. The frequency at which they appear (Hz) is divided by the main frequency of the instrument (MHz) and thus a universal scale is obtained, the δ scale or ppm (parts per million). The following considerations must be taken into account:
- Chemically equivalent protons have the same chemical shift.
- Carbons attached to electronegative groups are “unshielded” and tend to shift NMR signals from adjacent protons to “lower field” (high ppm values).
- Protons bound to oxygen or nitrogen have highly variable chemical shifts and are also sensitive to concentration, solvent, temperature, etc.
- The π-systems of alkenes, aromatic compounds and carbonyls strongly unshield the bound protons by shifting them to “low field” (higher ppm values).
- The zero of the spectrum is arbitrarily taken as the singlet signal given by tetramethylsilane (TMS; (CH3)4Si).
- Organic compounds give proton NMR signals in the range of 0-10 ppm usually and carbons in the range of 0-220 ppm.
Integral
The area of each signal responds to the number of hydrogens that are responsible for it.
Signal splitting in 1H-NMR (multiplicity)
The hydrogens close to each other influence each other and cause that the signals can be complex (doublets, triplets, quartets, quintets, etc…, multiplets). This multiplicity is due to the phenomenon known as “spin coupling” which originates from the interaction of the magnetic field of the proton with the electrons of the bond. In essence, each proton presents one of the two possible spin orientations under the applied magnetic field. Therefore, the magnetic field “felt” by an adjacent proton can have two possible values. The result is that n protons will split an adjacent proton into (n+1) peaks. The intensities of those peaks are simply the result of the possible spin orientations, the protons of a CH2 can have the possible spins:
↑↑ (↑↓ ↓↑) ↓↓
1:2:1
The central spins are degenerate, therefore, a proton adjacent to a CH2 undergoes a 1:2:1 splitting.
| No. of H responsible for splitting | Multiplicity (Multiplet relative intensity) |
| 1 | doublet (2) 1:1 |
| 2 | triplet (3) 1:2:1 |
| 3 | quartet (4) 1:3:3:1 |
| 4 | quintet (5) 1:4:6:4:1 |
| 5 | sextet (6) 1:5:10:10:5:1 |
| 6 | septet (7) 1:6:15:20:15:6:1 |
| … | … |
Mass spectrometry
We will summarize the key aspects of MS for structure determination in this section. MS gives the result of the ionization of the organic compound, showing the relative stability of the ions formed in that ionization and how they fragment.
The ions present with greater intensity always correspond to those that are more stable from the physicochemical point of view.
For example, a benzyl or allylic cation is more stable than a carbocation 3º > 2º >1º. Thus, the more electronic delocalization or resonant forms present, the more stable the structure of the ion.
Nitrogen rule
If a molecular ion (M+-) is odd, the compound contains an odd number of nitrogens.
McLafferty rearrangement
Olefins and other unsaturated compounds with an H hydrogen in the γ position with respect to the ethylene bond may undergo a process involving the breaking of the allylic bond and the transfer of the γ H hydrogen to the allylic double bond via a 6-membered cyclic transition state.
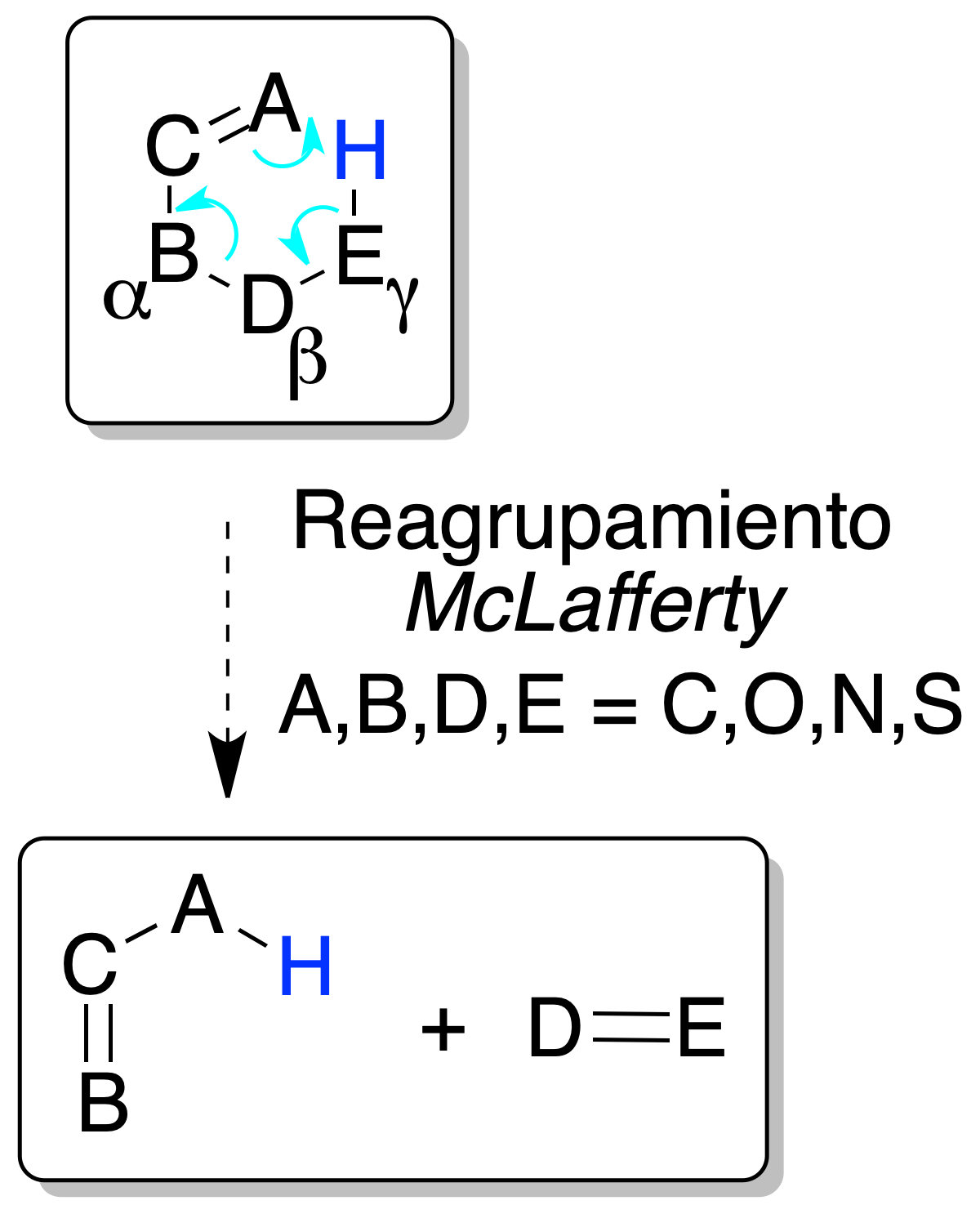
The charge of the cation remains within one or the other of the fragments: of the two ions, that of the more substituted (more stable) olefin predominates.
Examples
Full Professor of Organic Chemistry at the University of Granada, with a long-standing research career in Computational Chemistry and molecular modeling and design.