
Conversion of alkenes to alkanes
Catalytic hydrogenation
The addition of one mole of H2 to a double bond leads to the formation of a saturated compound (alkane). The conditions (temperature, pressure, etc.) of hydrogenation are relatively mild, the presence of a catalyst being essential, since the mixture of hydrogen and alkene does not react spontaneously due to the high activation energy of the process.
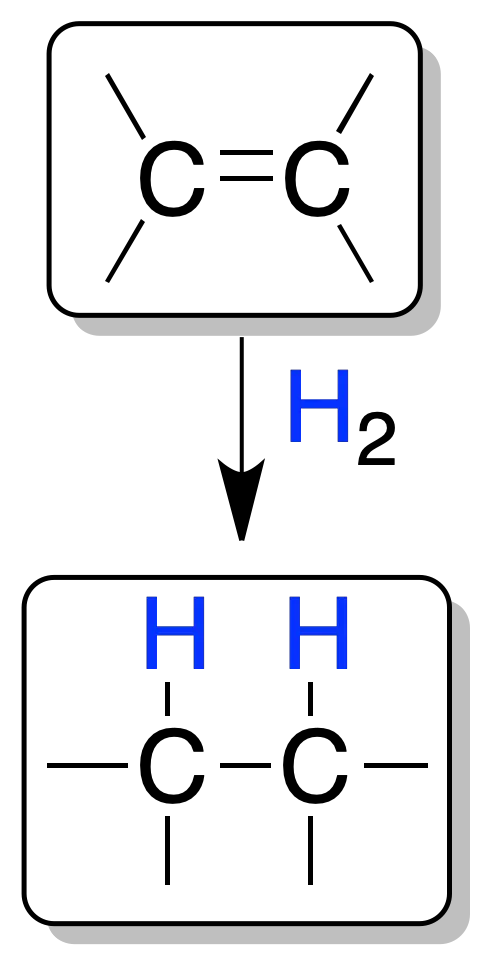
The most commonly used catalysts are transition metals (Pd, Pt, Ni, etc) on an inert support such as carbon or ionic salts. The most commonly used solvents for this reaction are ethanol, hexane or acetic acid.
Under these conditions the catalysts are totally insoluble in the reaction medium, so it is a heterogeneous catalysisphenomenon.
Catalysts soluble in organic solvents can also be used (homogeneous catalysis). They are usually metal complexes formed by a transition metal (Rh, Ru or Ir) with organic ligands, e.g. Ph3P.
The transformation is stereoselective (and can be stereospecific): the two hydrogen atoms that are incorporated into the molecule do so on the same side of the double bond (addition-syn).
Reduction to alkanes with borane
The reaction of borane with an alkene is a concerted and stereospecific process.
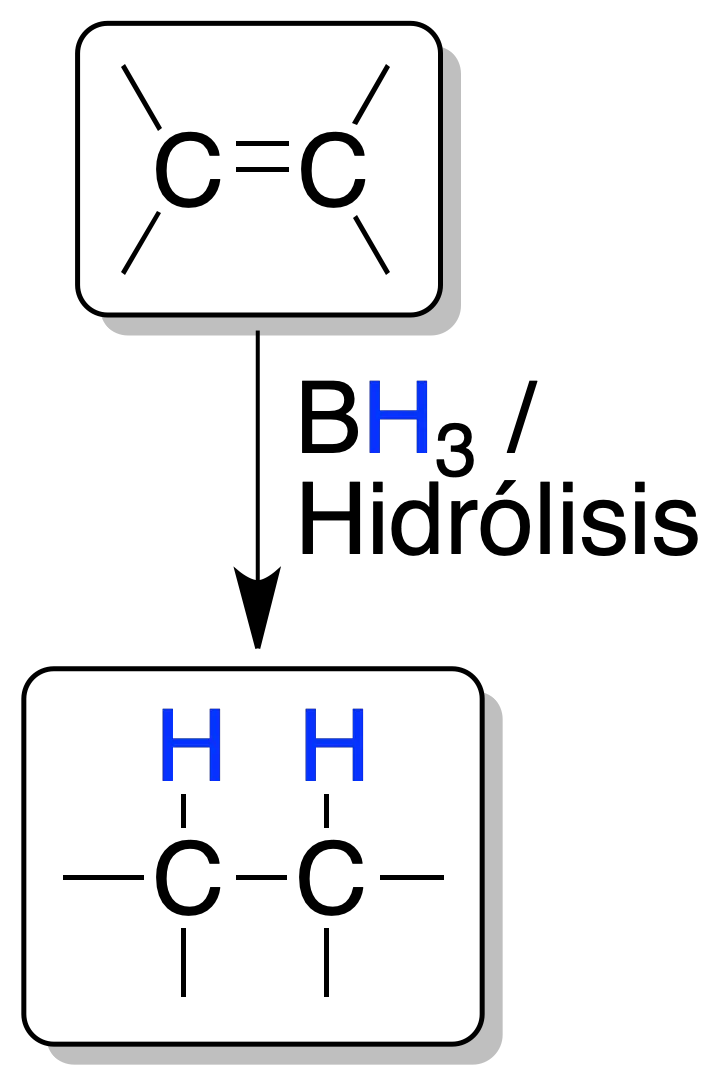
Boron is bonded to the least substituted carbon (see hydroboration-oxidation reaction). Hydrolysis with an aqueous acid solution of the alkylborane intermediate produces an alkane.
An alternative to borane is the use of 9-BBN.
Electrophilic additions to alkenes
Alkenes possessing the π electrons of the double bond are capable of reacting with electron-deficient compounds (electrophiles), depending on the nature of these compounds, different products will be obtained.
A summary of this reactivity is shown in Table 1.
| Reactive Electrophile | Type of attack (Stereo-selectivity) | |
| H2O / H3O⊕ | H-OH | Markovnikov (-) |
| BH3 / oxidation | H2B–H | Anti-Markovnikov (sin-addition) |
| Mercury(II) acetate | Hg(OAc)2 | Markovnikov (anti-addition) |
| Sulfenyl chloride | RS-Cl | Markovnikov (anti-addition) |
| X-H (sin iniciadores de radicales) | X-H | Markovnikov (-) |
| X2 | X-X | – (anti-addition) |
| X2 / H2O (XOH) | XOH | Markovnikov (anti-addition) |
Acid-catalyzed hydration
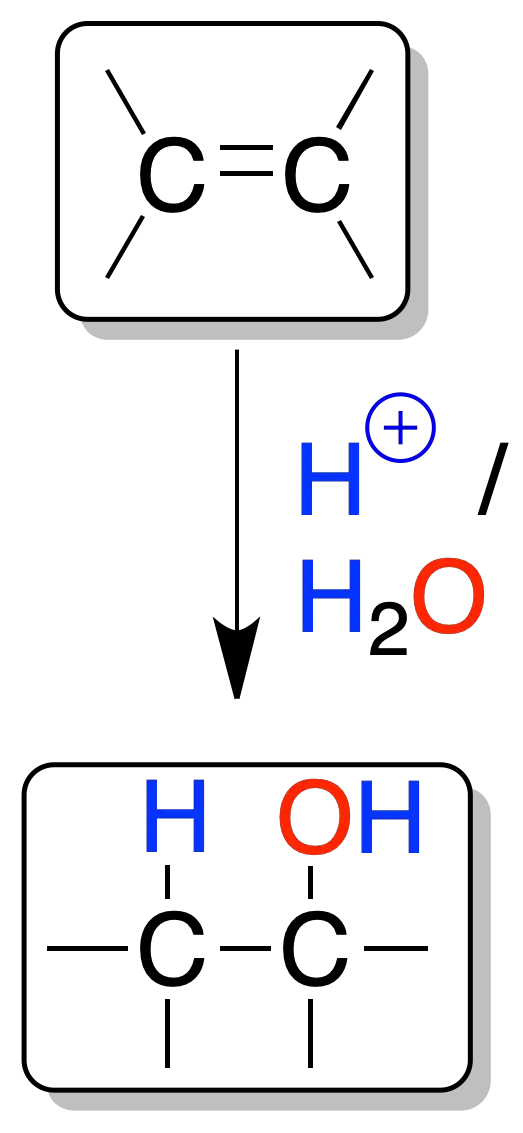
An alkene treated with an aqueous solution of an acid with a low nucleophilic anion (SO42-, PO43-) adds one molecule of water.
An initial protonation occurs, generating a carbocation, on which a water molecule subsequently attacks.
Due to the formation of carbocations, transpositions of the carbonate skeleton are sometimes observed.
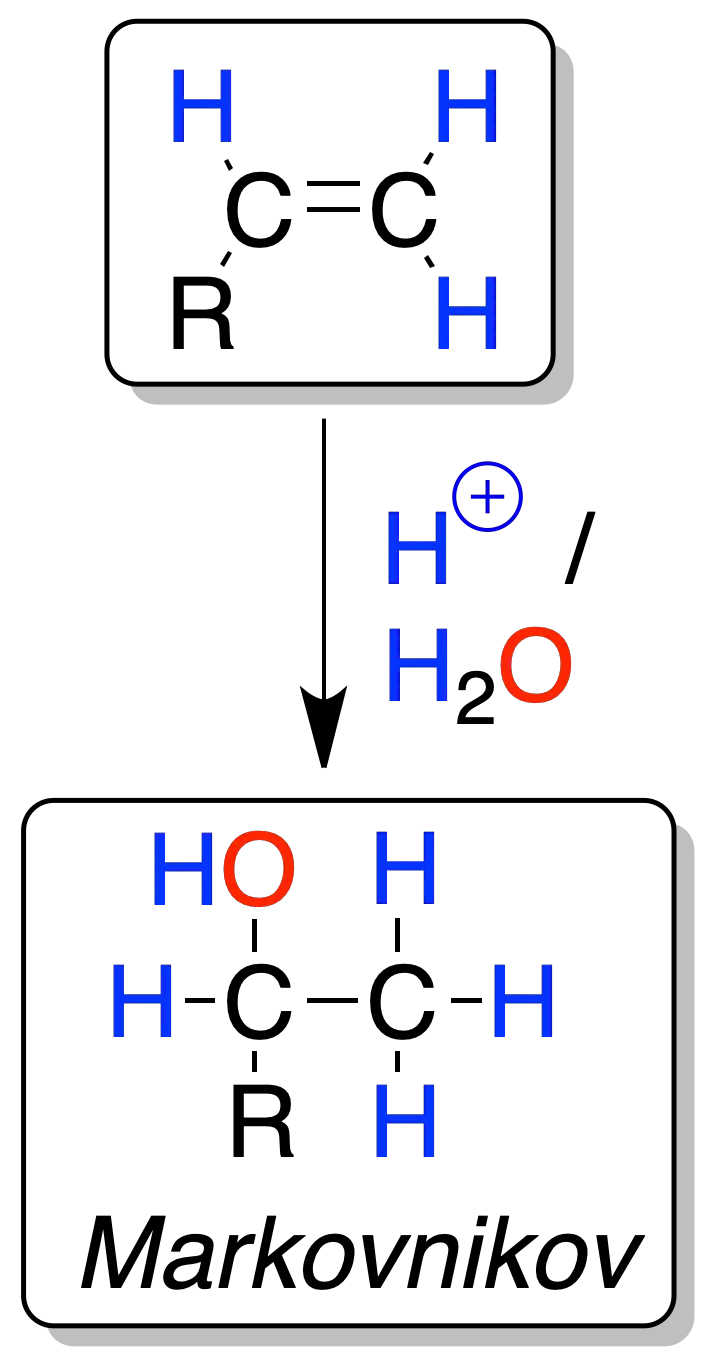
The reaction is regioselective since the H⊕ proton (electrophile) binds to the less substituted carbon and the HO⊖ group to the more substituted one.
This type of attack (in which the electrophile binds to the least substituted carbon and the nucleophile to the most substituted carbon) is known as Markovnikov’s rule.
This reaction reverts to the starting reagents by dehydration of alcohols.
The dehydration of alcohols is favored by hot and concentrated acids, while the hydration of alkenes is favored by excess water and low temperatures.
Concentrated sulfuric acid (H2SO4) can be added to some alkenes generating an alkylhydrogen sulfate.
The reaction is an electrophilic addition of the Markovnikov type.
When heated in water, alkynylhydrogen sulfate hydrolyzes readily, giving an alcohol (only in monosubstituted alkenes).
Polymers are produced in polysubstituted alkenes.
Oximercuriation-demercuriation
The alkenes react with mercury(II) acetate (H2O / THF) to give an intermediate (mercurial), which by reduction with NaBH4in basic media, generates an alcohol.
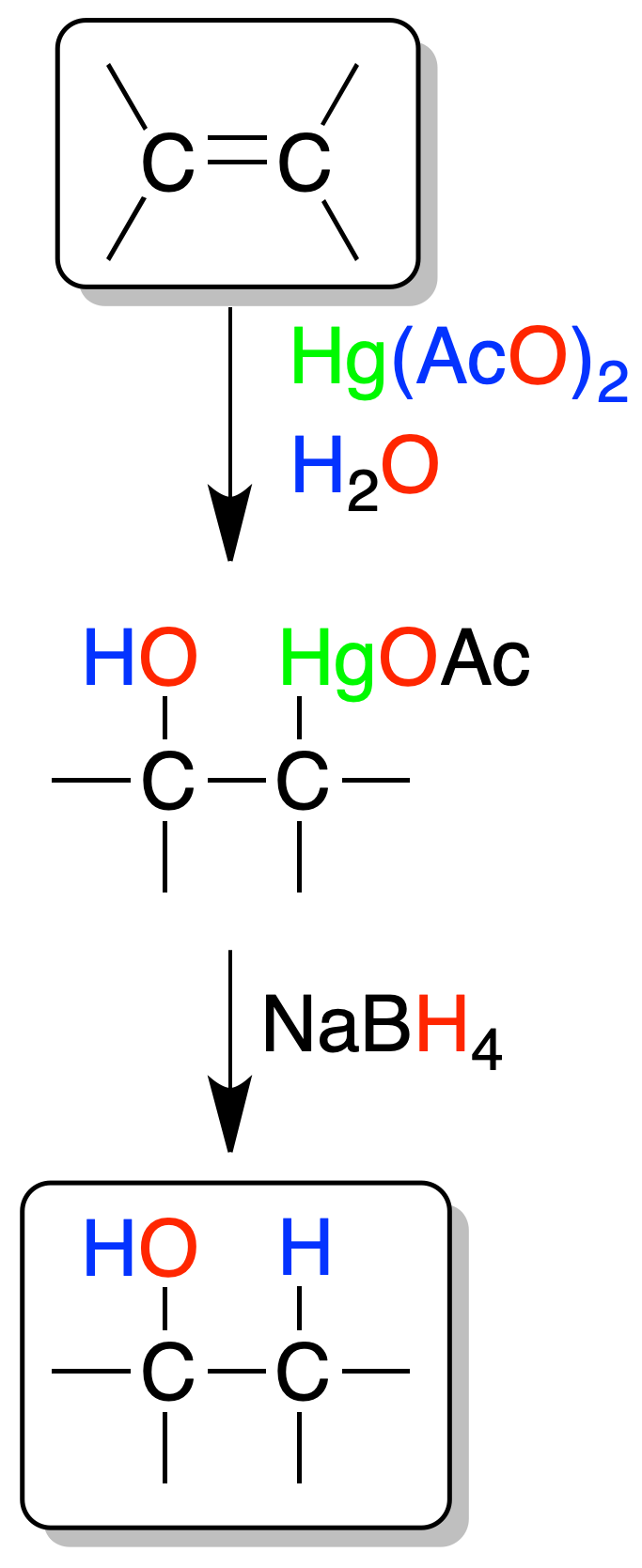
The regioselectivity is the same as that of the previous acid-catalyzed hydration, but has the advantage that no transpositions occur in it. Only a product of inverse regioselectivity to that of the hydroboration-oxidation reaction is obtained.
Mercury is bonded to the least substituted carbon, and an -OH to the most substituted (Markovnikov’s rule). Moreover, it is stereospecific, because the addition of mercury and hydroxyl is an addition-anti.
The reaction mechanism is explained by the attack on the double bond of the mercurinium ion ⊕HgOAc (dissociation of mercuric acetate). A cyclic complex is formed which subsequently opens from water, and ending with the loss of a proton. In the last reduction step with NaBH4, mercury is replaced by hydrogen.
Hydroboration / oxidation
It allows the addition of a water molecule to an alkene with regioselectivity opposite to the alkene hydration process seen in the previous section.
The process is a tandem of two reactions. First a trialkylborane is formed, which is not isolated, and subsequently, it is oxidized with hydrogen peroxide.
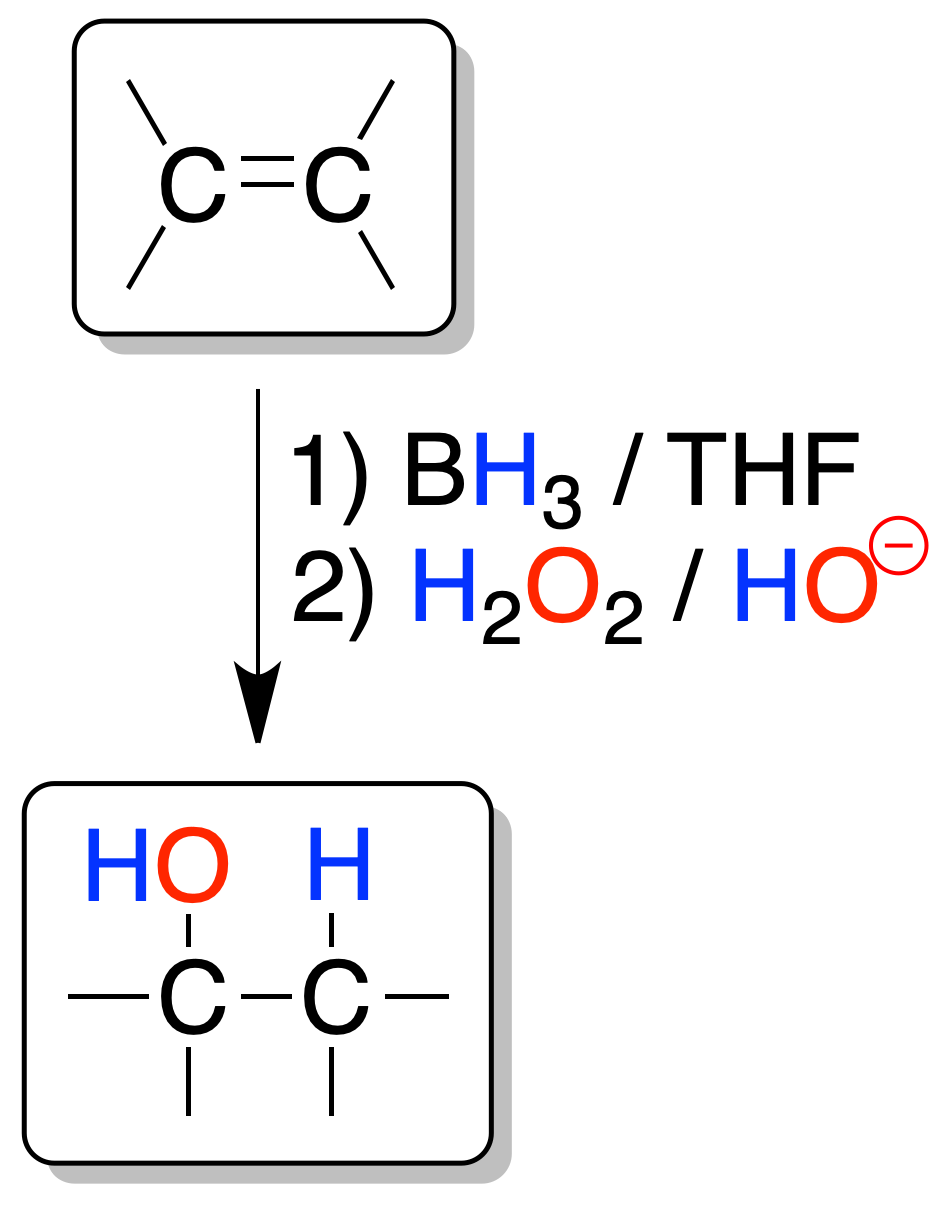
Each BH3 borane molecule can react with three alkene molecules. The reaction is regioselective, since boron is bonded to the least substituted carbon (the -OH group is generated from the alkylborane by oxidation of the latter) and hydrogen to the most substituted carbon (anti-Markovnikov).
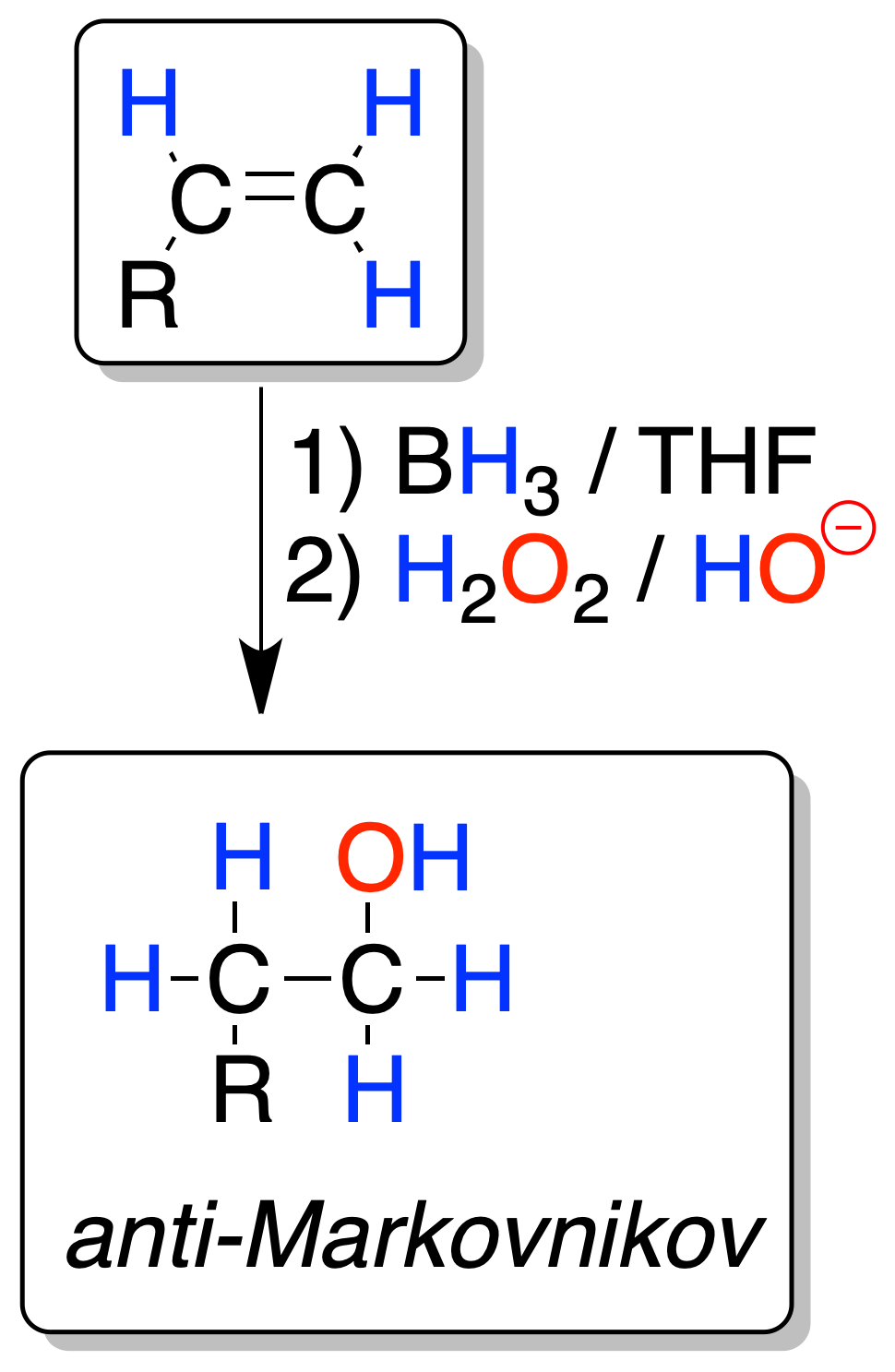
The reaction is also stereospecific, since the addition of the boron and the hydrogen atom occur on the same side of the alkene (addition-without).
Electrophilic HX addition (in the absence of radical initiators)
The reaction of an alkene with a hydrogen halide in the absence of radical initiators produces an alkyl halide.
The proton H⊕ bonds on the least substituted carbon and generates the more stable carbocation, which is attacked by the halogen found as X⊖.
The process follows Markovnikov’s rule, since the tertiary 3º and secondary 2º carbocations are more stable than the primary ones, and transpositions can occur.
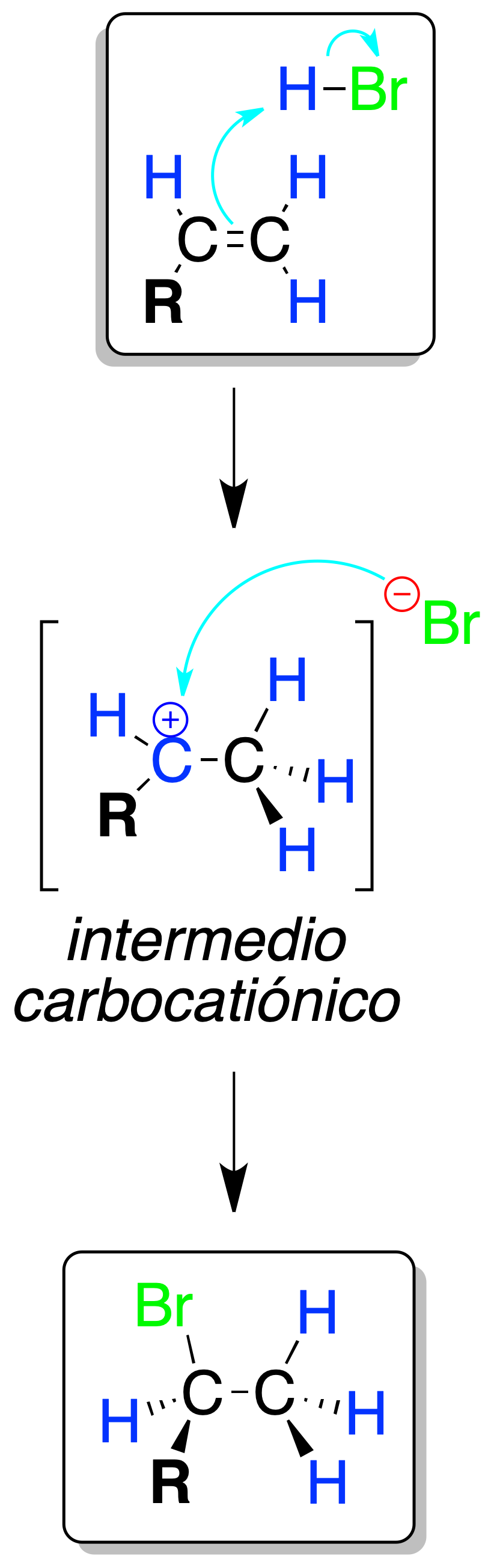
If nucleophiles other than X⊖ are present in the medium, mixtures of products are produced as a consequence of the attack of these nucleophiles on the carbocationic intermediate.
The reactivity of hydrogen halide with alkenes follows the order HI > HBr > HCl >> HF.
Moreover, performance is improved in the absence of light.
Addition of halogens
Chlorine and bromine are added with good yields to alkenes giving a neighboring dihalogenated compound.

The reaction rate increases with the polarity of the solvent, due to the polar character of the reaction intermediate.
The solvents used are aprotic solvents such as Cl2CH2, Cl4C, or Cl3CH or low nucleophilic protics (acetic acid).
The reaction intermediate is a cyclic halonium ion (chloronium, Cl⊕, or bromonium, Br⊕).
The addition occurs on different sides of the double bond (addition-anti) and is therefore stereospecific. Depending on the substituents and/or the stereochemistry of the starting alkene, mixtures of stereoisomers can be generated.
Addition of halogens to double bonds followed by double elimination of neighboring dihalides leads to the formation of alkynes.
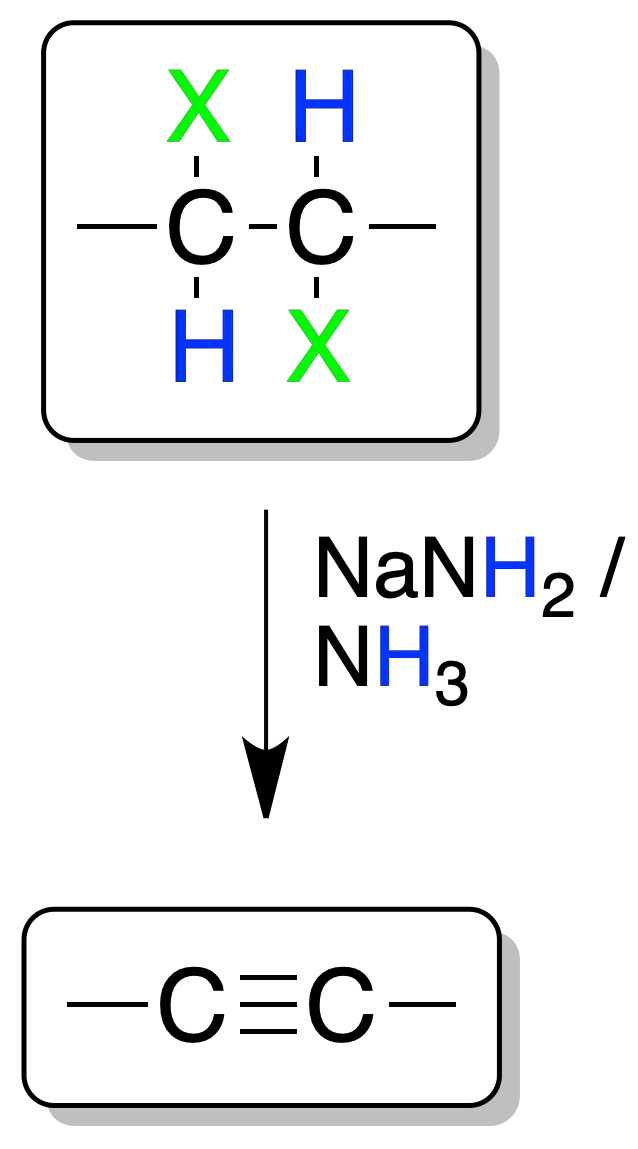
For the elimination reaction to occur, the use of a strong base such as NaNH2 in NH3 is necessary. The reaction occurs only on mono- or di-substituted alkenes.
Halohydrins formation
In aqueous solution, chlorine (Cl2) and bromine (Br2) react with alkenes to give halohydrins (α-haloalcohol).
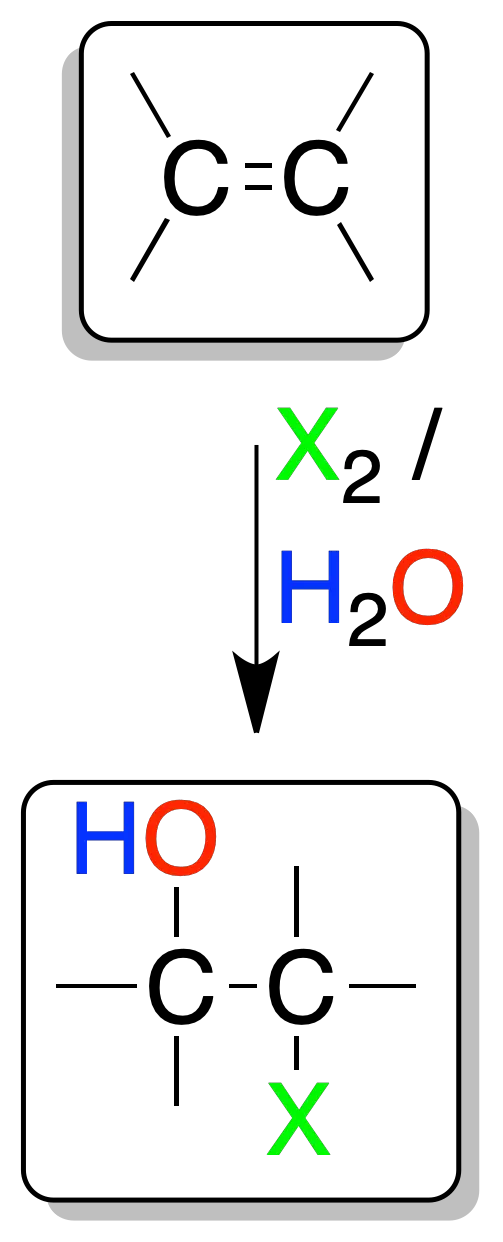
These compounds are synthetic intermediates useful for the preparation of epoxides.
An intermediate halonium ion is formed, which is attacked by water on the opposite side to bromine (addition-anti).
Moreover, the water attack is regioselective: it occurs on the most substituted carbon (with the highest positive charge density), thus a Markovnikov-type addition.
Electrophilic sulfenyl chloride addition
The reaction of sulfenyl chloride (RSCl) on olefins produces chloroalkylsulfides. The reaction is regioselective.
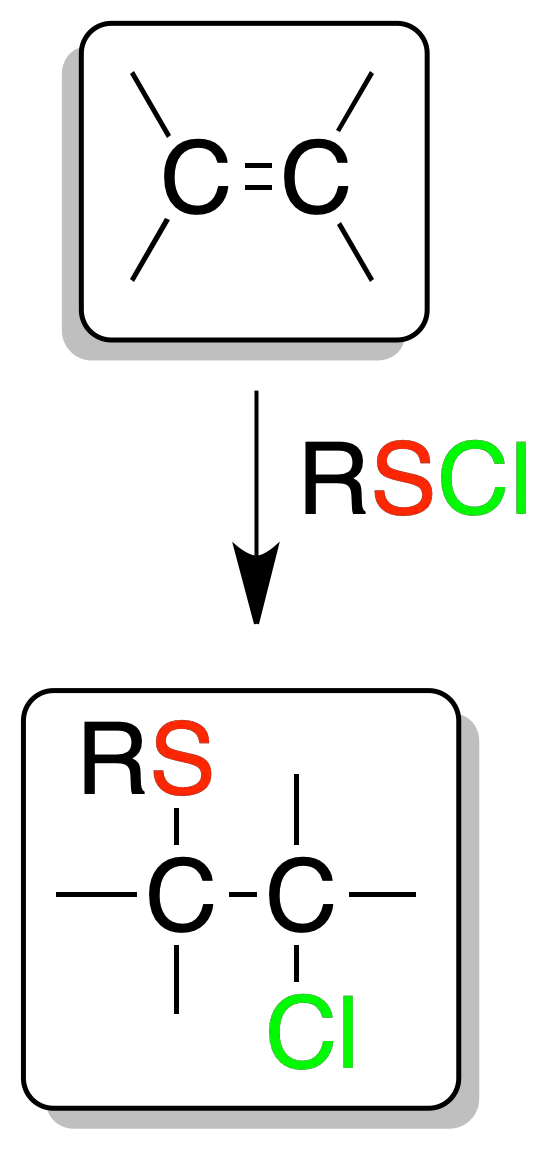
Sulfur is fixed on the least substituted carbon (Markovnikov) and is also an addition-anti.
On the other hand, chloroalkylsulfides are synthetic intermediates for obtaining epoxides.
Other additions to alkenes
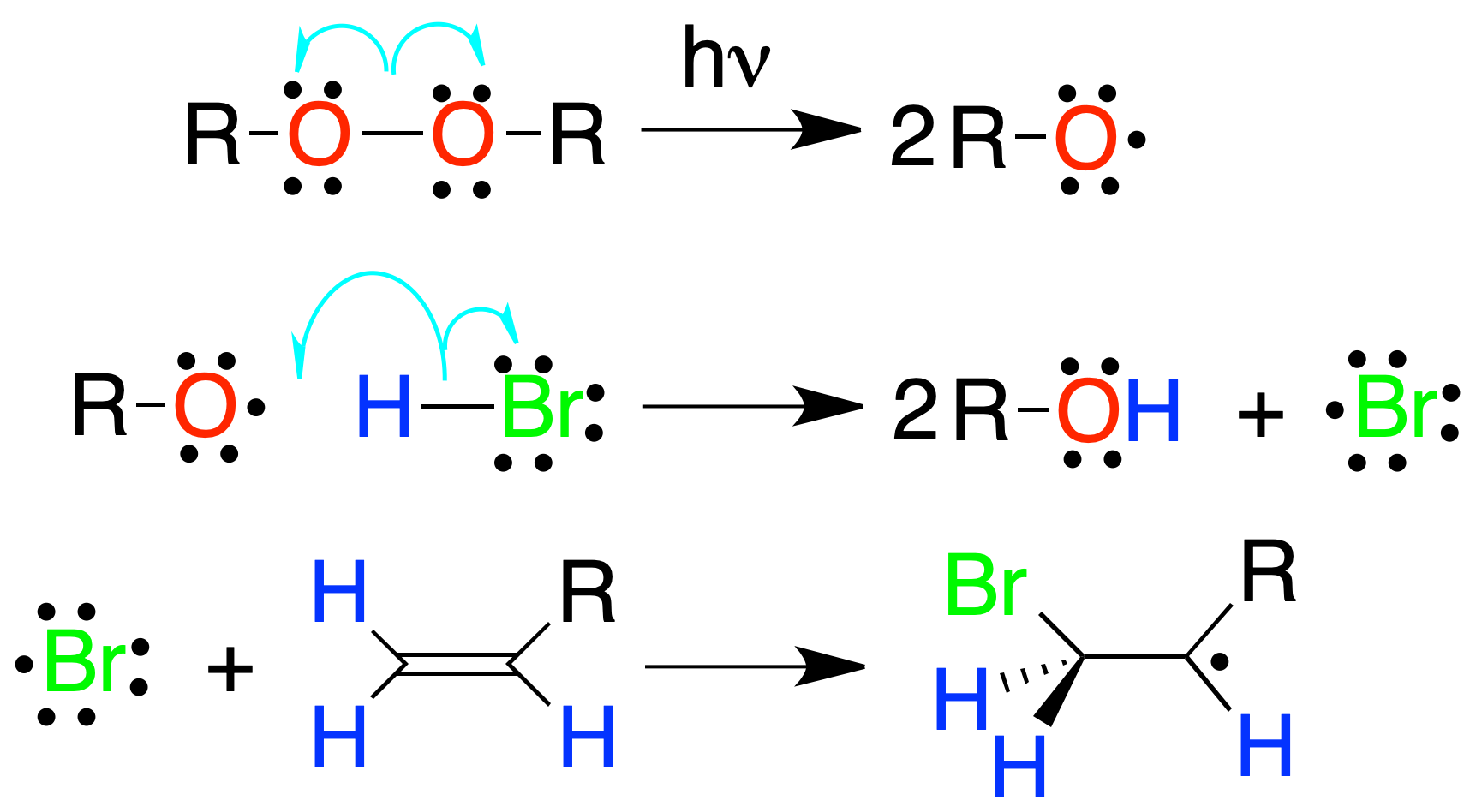
Radical addition of HBr
The addition of HBr to alkenes in the presence of an initiator such as peroxides, light or heating leads to an alkyl bromide with an inverse regioselectivity (anti-Markovnikov) to that of the electrophilic HBr addition reaction, because the more substituted (more stable) radical is produced.
Addition of carbenes
Carbenes are added to double bonds generating cyclopropanes. The addition of a carbene to a double bond is stereospecific, occurring on one side of the double bond. In addition, carbenes are highly reactive divalent synthetic intermediates (RR’C:).
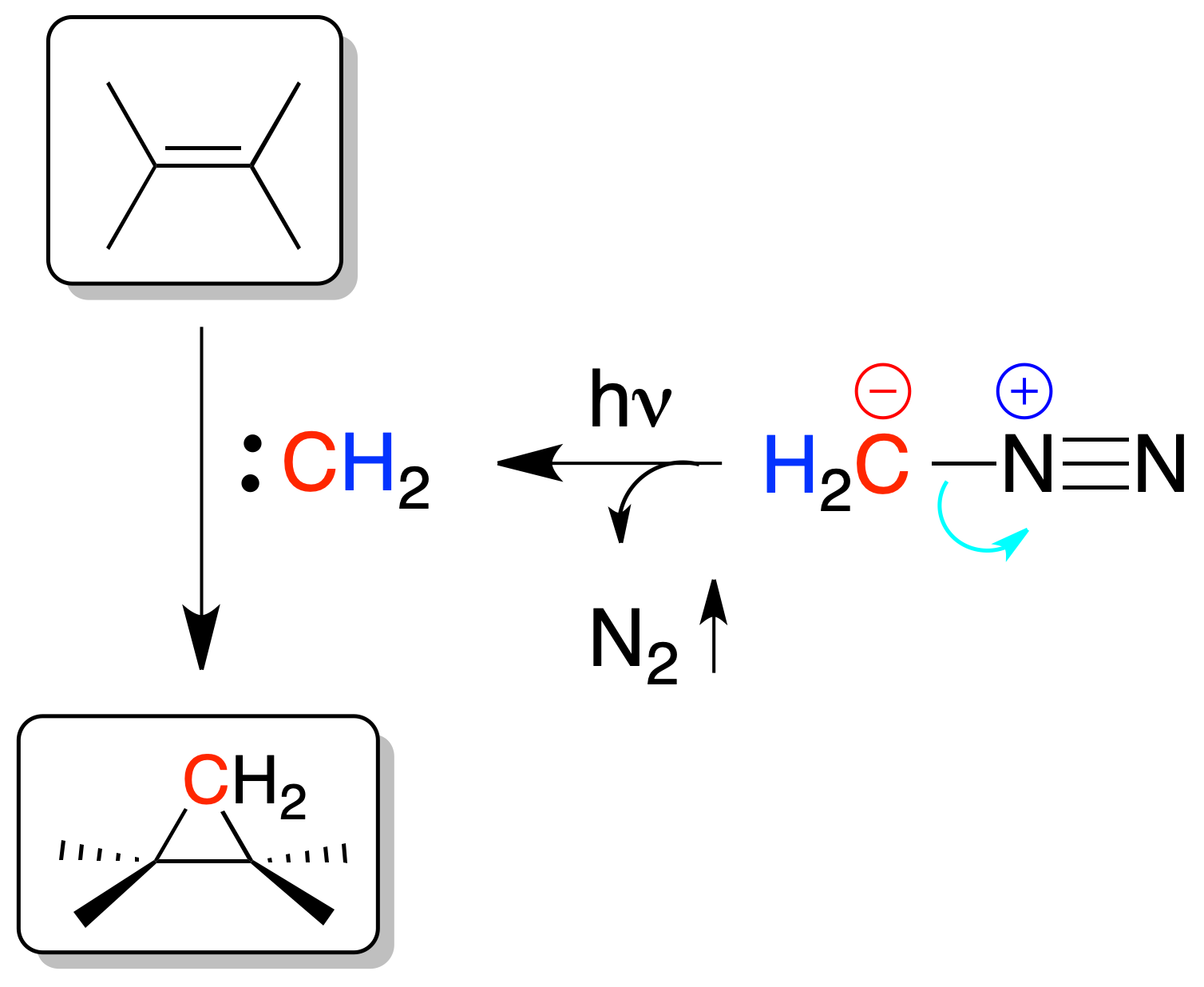
They can be generated from a diazoalkane derivative with light or heat, from chloroform in a strongly basic medium, or by the Simmons-Smith reaction.
Oxidation of alkenes
The oxidation of alkenes produces different compounds depending on the oxidant and reaction conditions used.
Epoxidation
The treatment of an olefin with a peracid allows the synthesis of an epoxide or oxirane.
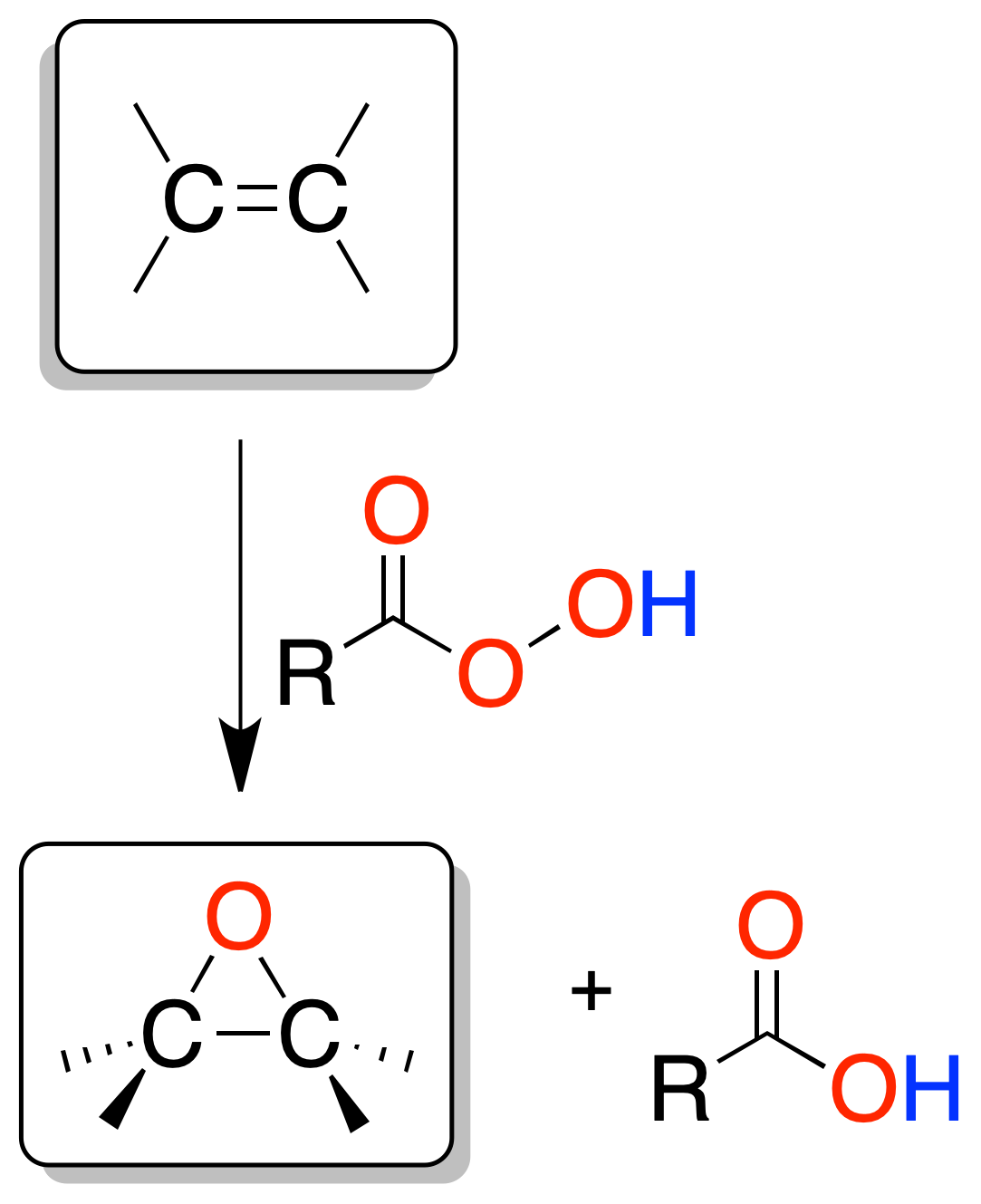
The reaction is a concerted process resulting in a syn-type addition. The most commonly used peracetic acid is peroxyacetic acid (peroxyacetic and m-chloroperbenzoic acid) and the solvent, acetic acid.
Other solvents may be Cl2CH2 or CHCl. The relative stereochemistry of the substituents of the double bonds (Z or E) is maintained in the epoxide.
Oxidation of alkenes to vicinal diols (glycols)
The vecinal diols (glycols) can be obtained from alkenes by oxidation with MnO4⊖ permanganate in aqueous medium (pH > 8, basic conditions) at low temperature, or with OsO4 in aqueous sodium bisulfite solution. In both cases, a five-membered cyclic intermediate (cyclic osmate) is postulated.
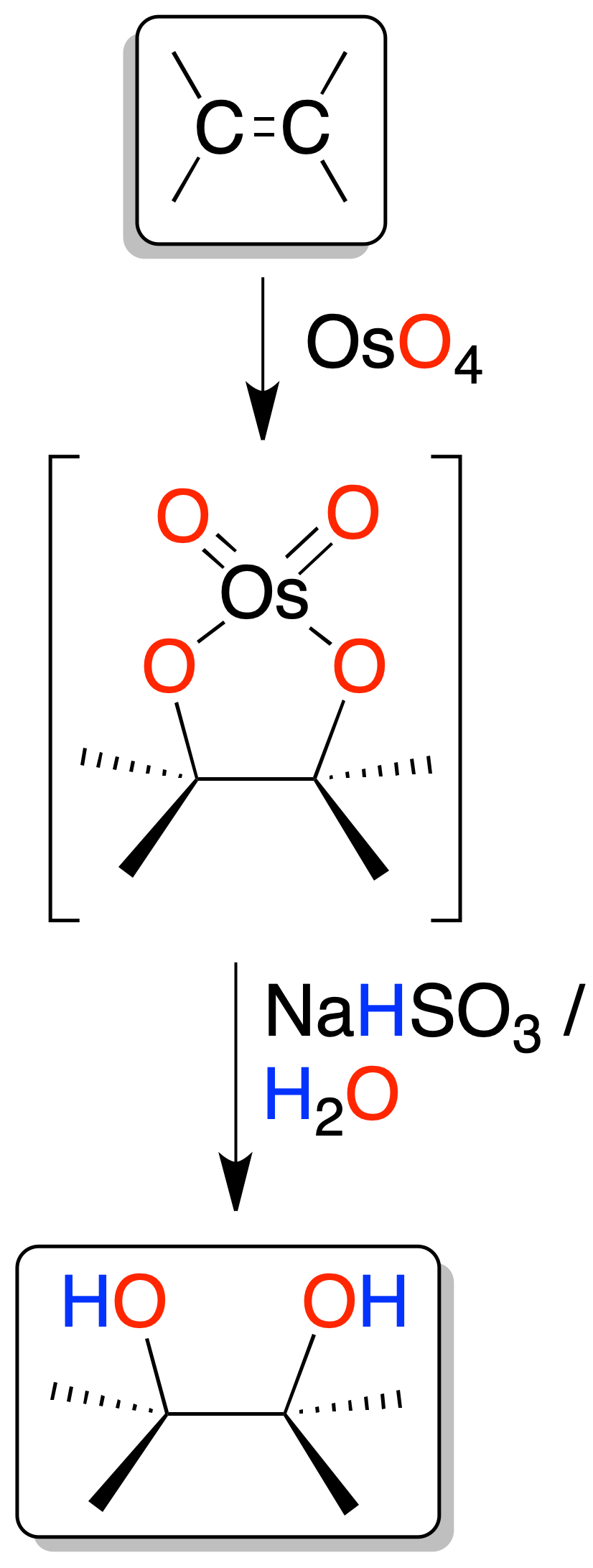
Osmium oxide OsO4 can be used in catalytic quantities in the presence of an oxidant, tert-butylhydroperoxide or hydrogen peroxide (H2O2), so as it is consumed it is regenerated in situ.
Ozonolysis
Ozonolysis (reaction of alkene with ozone) on alkenes also produces an oxidative cleavage of the double bond, but unlike the previous case, aldehydes or formaldehyde are obtained. If the desired oxidation product is aldehyde instead of carboxylic acid, we will choose ozonolysis.
The reaction intermediate is called ozonide, and its hydrolysis produces mixtures of carbonyl compounds: aldehydes and ketones. Treatment with a reductant (Znº in acetic acid or dimethyl sulfide) allows isolation of the reaction products.
The intermediate ozonide is explosive, so it must be worked at low temperatures.
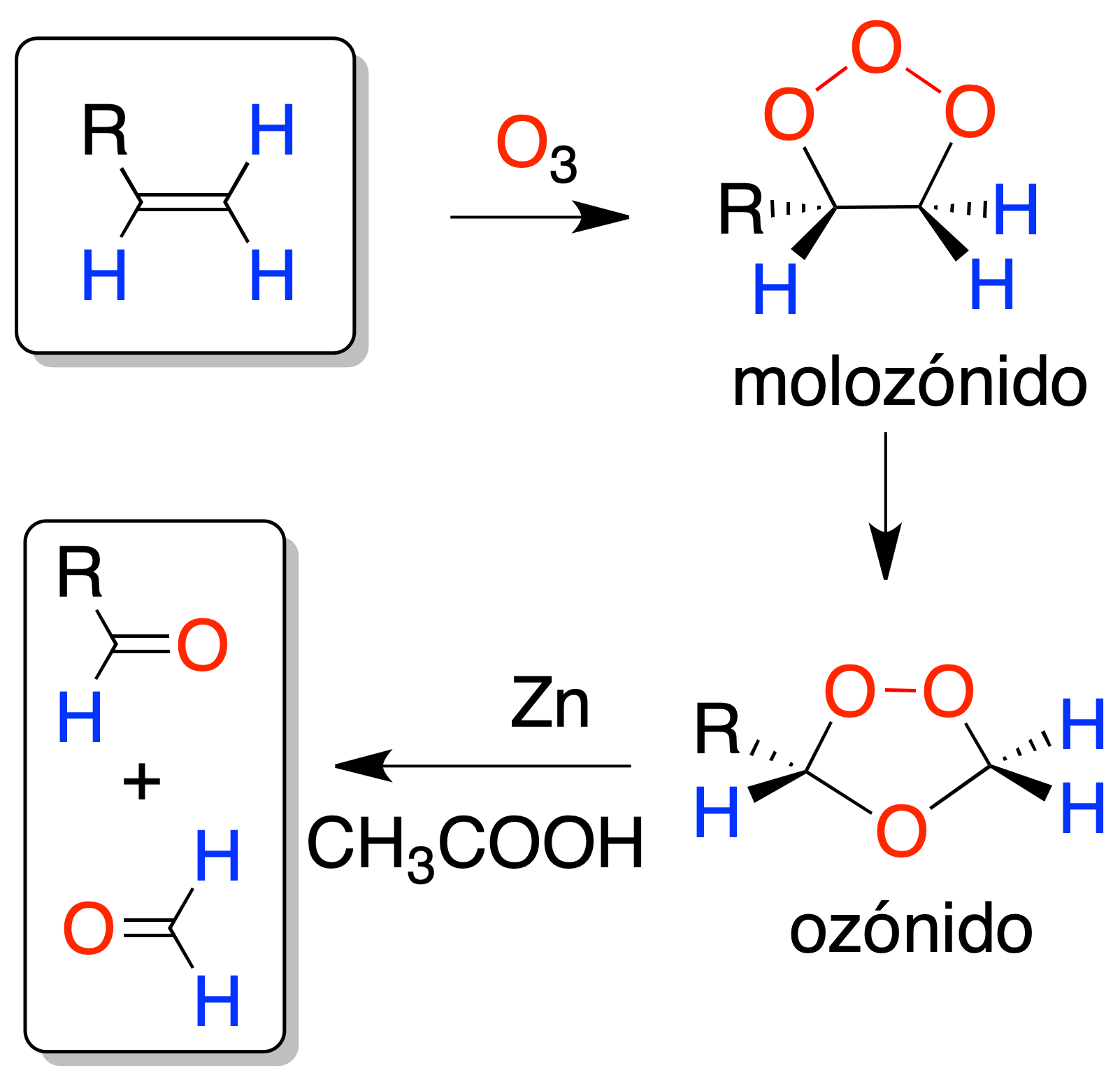
Both in this case and in the following, by analyzing the fragments, it is possible to determine the position of a double bond in a molecule, although not its stereochemistry.
If one of the substituents of the double bond is hydrogen, at least one of the carbonyl compounds obtained would correspond to an aldehyde, but if the double bond is at the end of a chain (terminal double bond), formaldehyde would be obtained.
Oxidative cleavage with permanganate
In permanganate oxidation, if the temperature or pH is not controlled (high temperatures or acidic or neutral conditions) there is a danger of over-oxidation (oxidative cleavage of the double bond), to give carboxylic acids or ketones, or carbon dioxide (in the case of a terminal alkene).
As in ozonolysis, the analysis of the fragments allows us to know the location of the double bond.
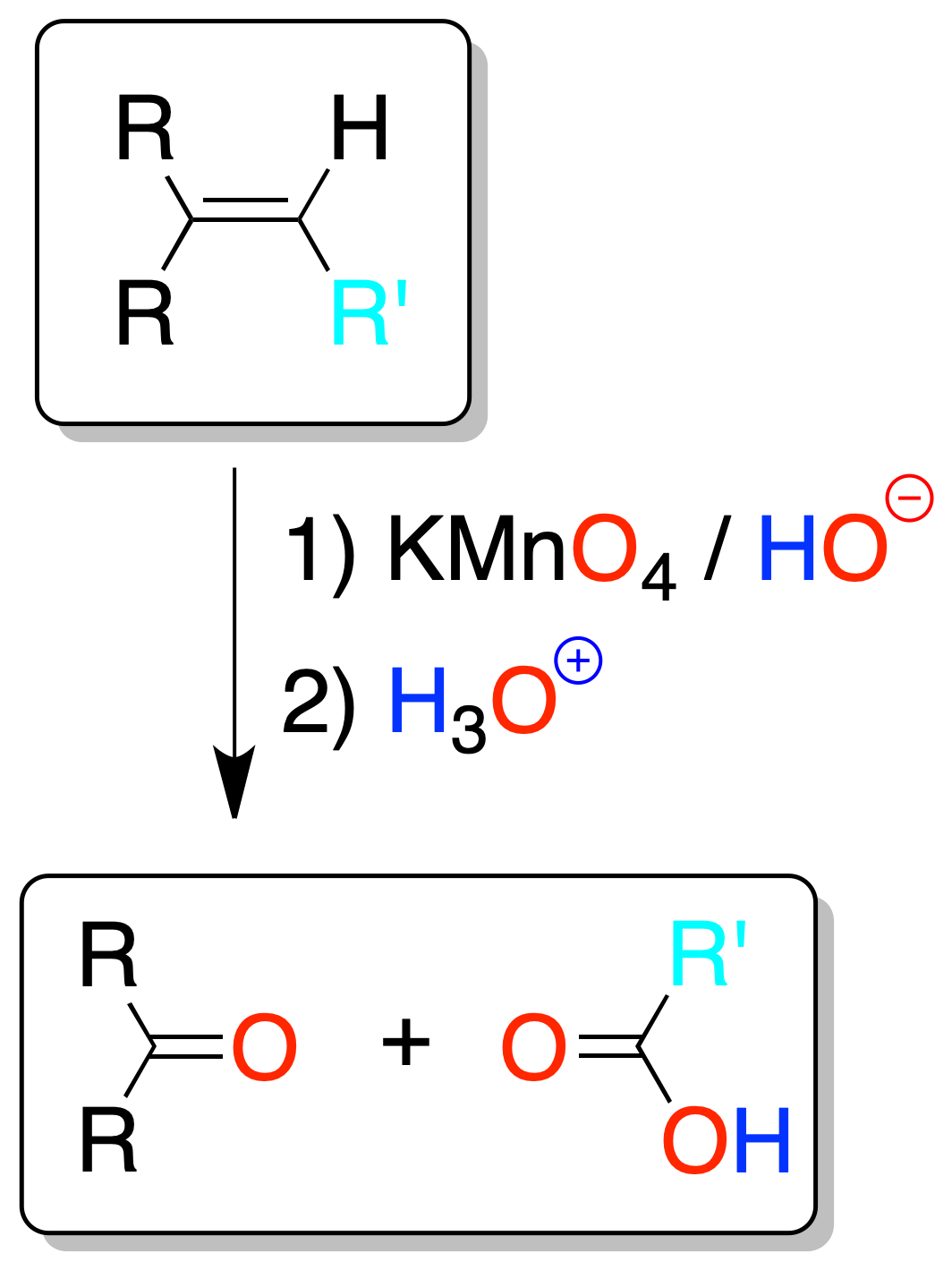
If a ketone is formed one of the carbons forming part of the double bond has two substituents. If CO2 is given off, the double bond would be at the end of a chain.
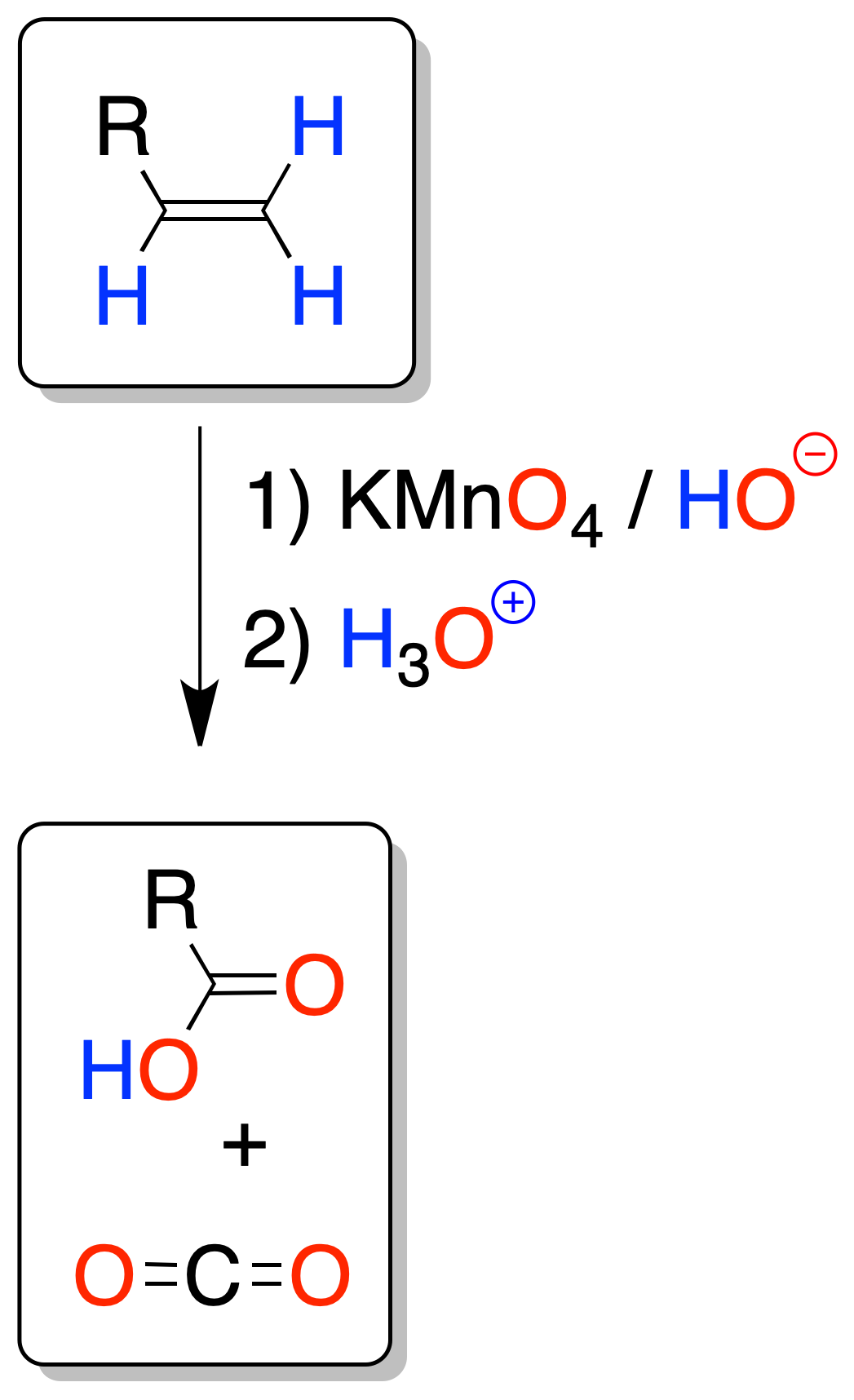
In order to isolate the acids obtained it is necessary to acidify, since the reaction is carried out in a basic medium forming carboxylate ions.
Alkene substitution reactions (allylic halogenation)
The presence of a double bond conditions the reactivity in positions close to it, so that new chemical properties appear.
In the vicinity of a double bond, two types of hydrogens can be distinguished: vinyl and allylic.
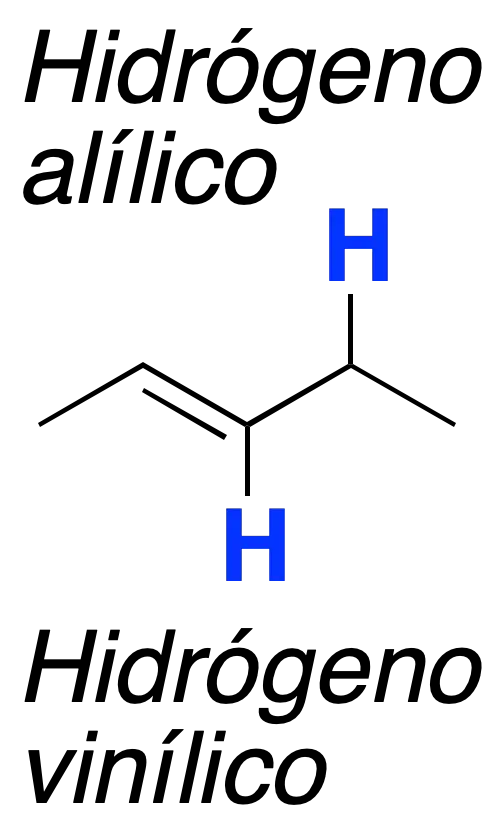
Particularly useful are the radical halogenation reactions that occur in the allylic positions of the double bonds, favored by the stability of the intermediate allylic radical.
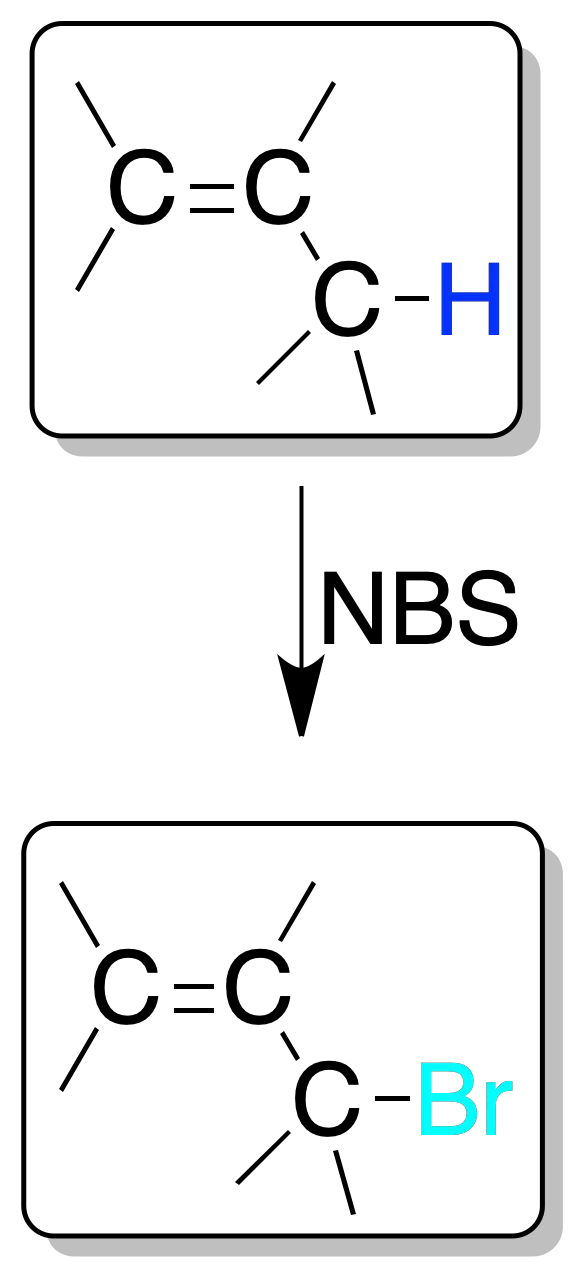
Halogenation in this position can be performed with Cl2 or Br2 by heating or irradiation with light of suitable frequency.
Bromination is usually done with NBS, which is a source of low concentrations of bromine radicals.
The reaction is of synthetic interest when the hydrogens in the allylic position are equivalent.
Polymerization of alkenes
Polymers are macromolecules (with high molecular masses, > 1000 amu) obtained by joining many smaller molecules together, which are called monomers.
Most natural or synthetic polymers come from the bonding of monomers that are alkenes. There are three types of polymerization:
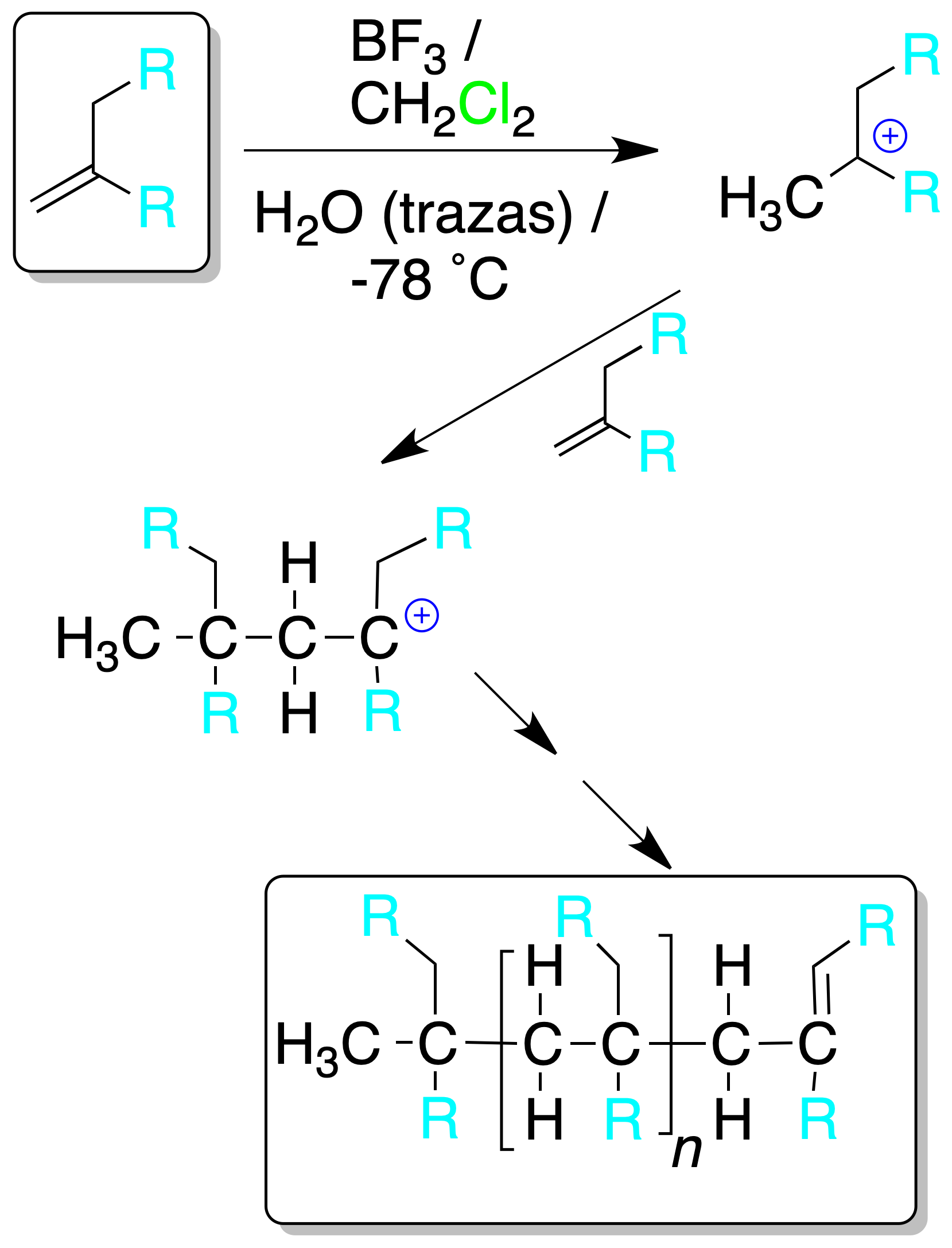
Cationic polymerization
Treatment of an alkene with strong acid (Brønsted’s acid in aqueous medium or Lewis acid with traces of water) generates a carbocation, which acts as an electrophile on a new alkene molecule.
The process continues repeatedly until a chain is obtained that stops growing due to the loss of a proton or the addition of a molecule for this purpose.
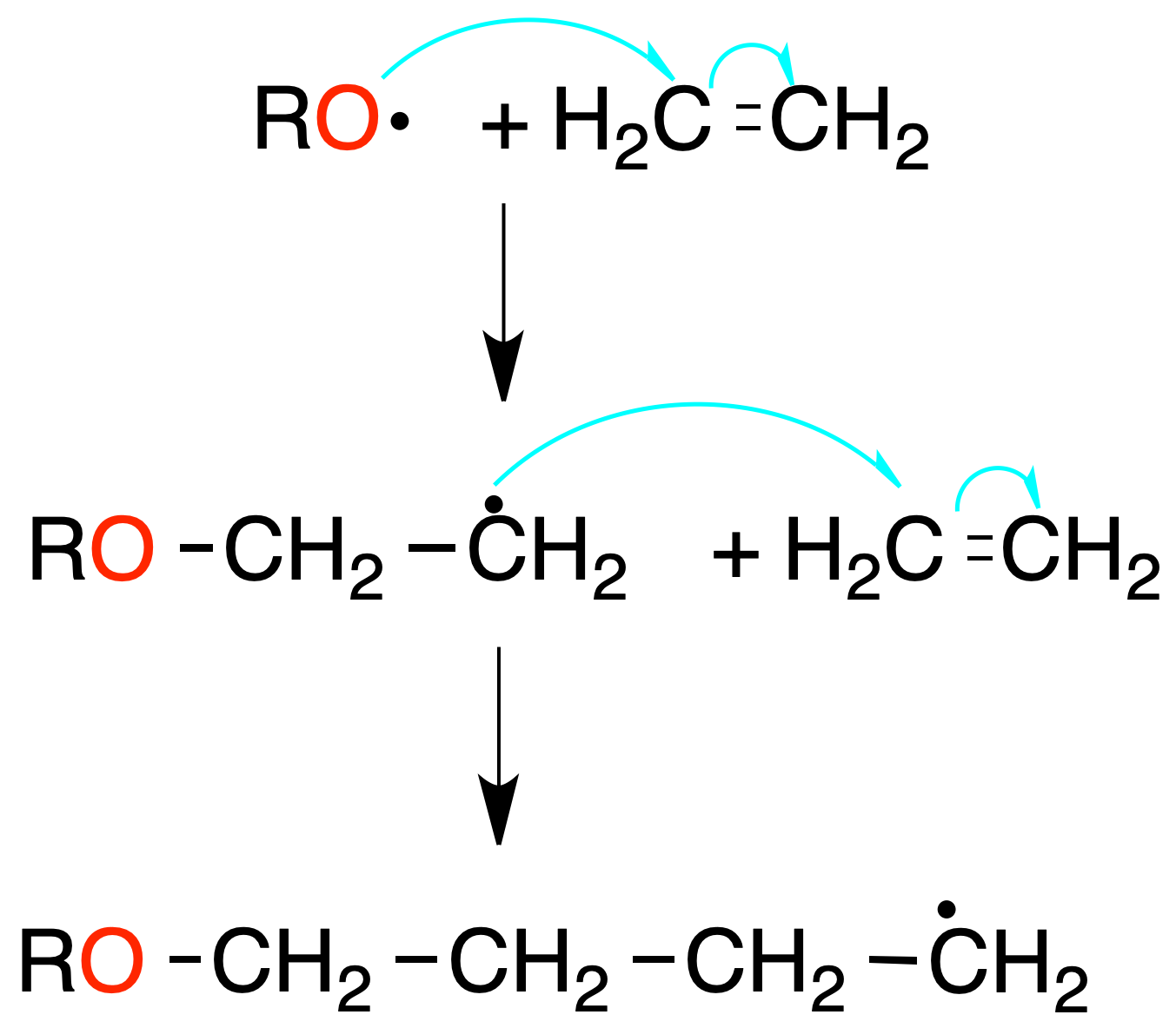
Radical polymerization
The monomer molecules react in the presence of a radical initiator (benzoyl peroxide or AIBN). The initiator generates a radical that attacks a monomer molecule, producing a new radical (initiation stage).
The radicals produced in this stage attack new monomer molecules (propagation stage) until a chain is formed. Polymer growth is terminated by combination of radicals, or disproportionation reactions (H· transfer, with formation of an olefin and a saturated compound).
Anionic polymerization
An anion is generated from the monomer by treatment with a strong base such as LiBu, and this anion attacks another monomer molecule. The process continues until the addition of a molecule that provides protons and prevents further chain growth (quench).
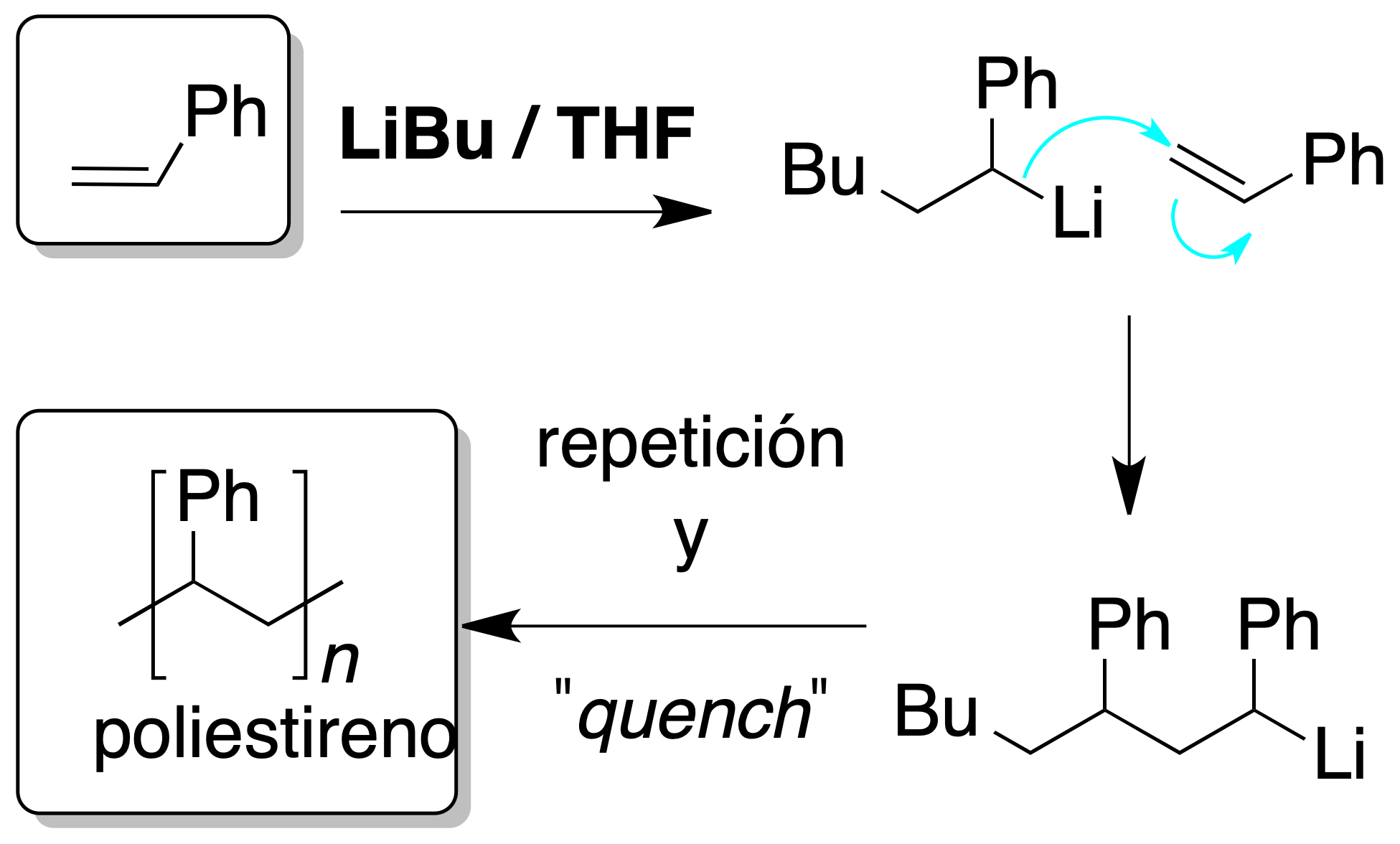
It is essential for the reaction to take place that the substituents of the double bond are able to stabilize the negative charge.
Back to the Synthesis and Reactivity of Organic Compounds.
Full Professor of Organic Chemistry at the University of Granada, with a long-standing research career in Computational Chemistry and molecular modeling and design.