
Introduction
In the following, pericyclic reactions are described from an applications point of view. That is, we will see practical examples of these reactions aimed at demonstrating that pericyclic reactions occur and preferentially versus the non-pericyclic alternative. Moreover, it is approached from an experimental and not a theoretical point of view. We try to obtain evidence justifying the existence of pericyclic reactions from an experimental point of view.
Pericyclic reactions are difficult to study experimentally since the reaction intermediates cannot be isolated. Therefore, what we will do experimentally is to prove that the non-concerted alternatives do not occur, for example by demonstrating that the reaction does not occur via a radical pathway or that it does not occur via an ionic intermediate, etc.
In addition, the stereochemistry of these reactions is usually studied experimentally, because they must be stereospecific. If a mixture of reaction products occurs when a single stereoisomer should occur, it is a clue that the reaction does not proceed by a pericyclic mechanism.
Cycloaddition reactions
We start with cycloaddition reactions that give rise to different cycle sizes.
Formation of three-membered rings
To obtain three-membered cycles one of the components has to act as a single atom through an ω-type interaction. The simplest reactions that generate 3-membered cycles are reactions with carbene (in its singlet or triplet state). Reactions in the triplet state cannot be pericyclic reactions since it is a radical and therefore will act through a radical-type reaction. But in singlet carbenes the electron pair can be represented as a pair of electrons on an sp2 orbital, with two additional substituents.
So experimentally it is observed that if the reaction is carried out in the presence of light which can act as a triplet, the reaction gives mixture of reaction products with the stereochemistry of the products, which shows that it proceeds by a radical mechanism. But if it is carried out in the absence of radicals the stereochemistry of the double bond is maintained, if it had a certain stereochemistry it will be maintained in the three-membered cycle. If it is a 4-electron system one of them has to act antarafacially, which is not difficult in a carbene, since such an interaction can occur:

That it will be an antarafacial interaction on carbene with a very simple geometry, examples are known in which the stereochemistry of the double bond is preserved and are therefore thought to be pericyclic type reactions in which the carbene acts antarafacially.

Chelotropic reactions
Many other examples are known in which the stereochemistry of the reaction is preserved, they are usually cycloreversion reactions instead of cycloaddition, e.g. by heating this type of structures cycloreversions are produced:
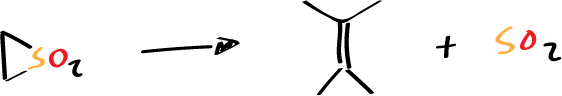


This double bond is produced and the stereochemistry of the reaction is preserved. This makes us think that they may be pericyclic type reactions, but since we cannot talk about the stereochemistry of the SO2, CO or SO group that is formed, it cannot be explicitly demonstrated that it is a pericyclic reaction.
Formation of four-membered rings
To form four-membered rings by cycloaddition reactions, the first thing we think of is π2+π2 reactions, but we know that this is thermally forbidden, however photochemically it can occur, so there are very simple methods of forming 4-membered rings.

However, some thermal reactions were found that presented interesting characteristics that at first made us think that they were pericyclic reactions and that later it has been demonstrated that they are radicalic and that are detailed since they show us tools to be used to demonstrate if a reaction mechanism goes by one mechanism or another.
In this type of reactions, the cycloaddition of dienic systems occurred but by 2+2 cycloaddition reactions, i.e., one of the two double bonds acted or also reactions of dienic systems with olefins substituted by halogens.
Specifically, the chlorinated and fluorinated 1,1-difluor,2-2 dichloro ethylene derivatives with butadienes have been studied. It was found that they were obtained from the possible reaction products, exclusively one against the other. That is, if we start from a 2+2 cycloaddition reaction and 1,2-butadiene was reacted:
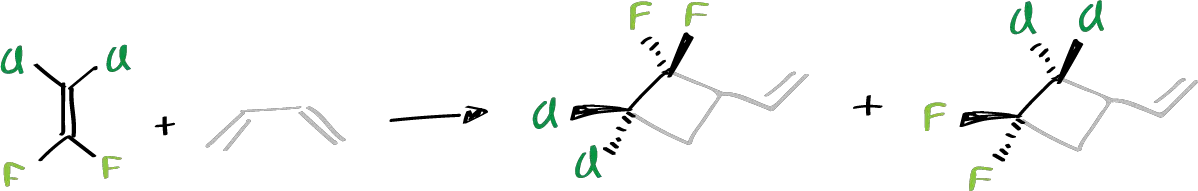
Of the two possible orientations of formation of the cyclic system, only the second one was obtained. In addition, if the diene system had a given stereochemistry (substituted dienes), and presents a cis or trans stereochemistry on the double bonds, the reaction product retains the stereochemistry of the double bond.
For example, if this system is reacted with these two compounds, one compound is formed from one and a second from the other.
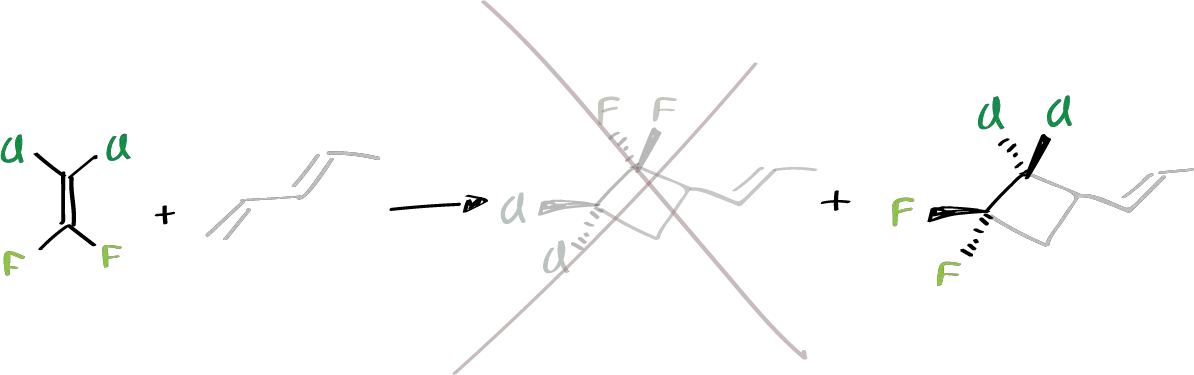
These features on the stereochemistry of the reaction and on the regiochemistry that occurs in the first reaction, was what led to think that it could be pericyclic type reactions that violate the selection rules, since it would be a case of a 2+2 supra–supra reaction.
Then, in a more exhaustive way, the stereochemistry of these reactions was studied, looking at all the possible reactions that occur against different dienes, so that they were made to react with these compounds:
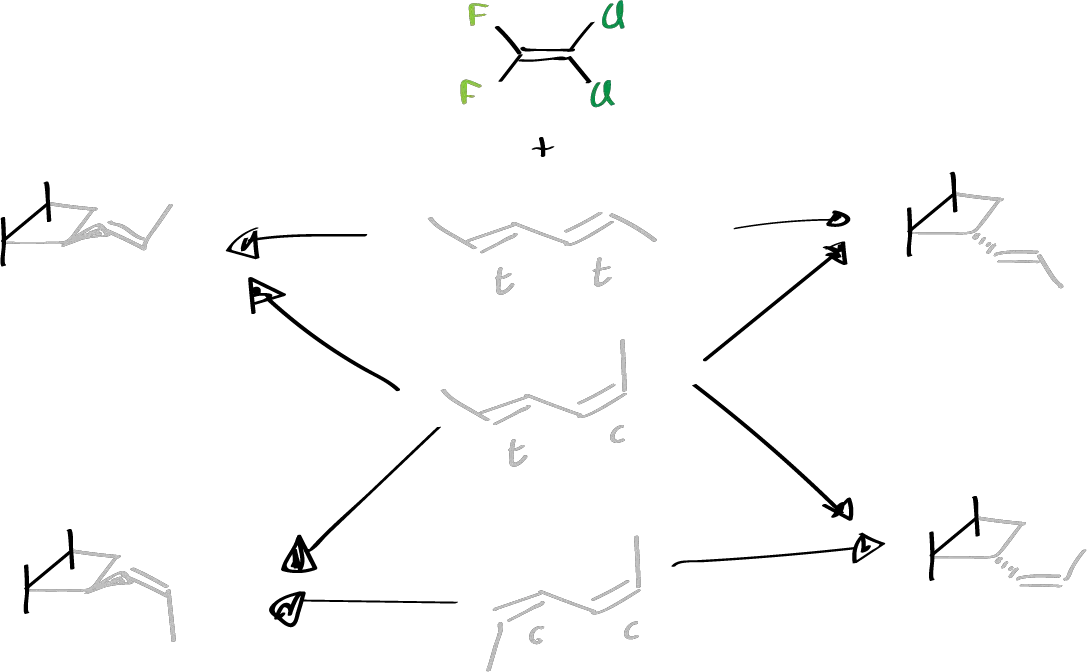
It was observed that from this same compound a mixture of the products was obtained, and this mixture corresponded to the trans isomer but with the upward substituent and the other product was the one with the downward substituent, which shows that there can be cycloaddition in both forms.
When the reaction was carried out with the second one, a mixture of 4 products was observed, two of them were those obtained previously and the other two are those with the substituent in the cis-form.
This indicates that the reaction is not stereospecific since from each one of them a single product should be obtained and a mixture of two is obtained and from the second one two should be obtained, one with a cis configuration and the other with a trans configuration. However, a mixture of the four is obtained.
All of the above led to think that it is a reaction with peculiar stereochemical characteristics, but it is not stereospecific and therefore cannot be a pericyclic reaction.
It is thought to be a radical reaction whose mechanism in the case of difluoro-dichloro is a stepwise reaction. First, the attack of this double bond on one of the two double bonds of the dienic system takes place. In addition, a radical attack takes place (a diradical originates). So in a first step we would obtain a radical which, due to the presence of that double bond, is an allylic radical that makes the rotation around that double bond impossible. However, along the next bond there can be rotation that evolves to the isomer that has the substituent down.
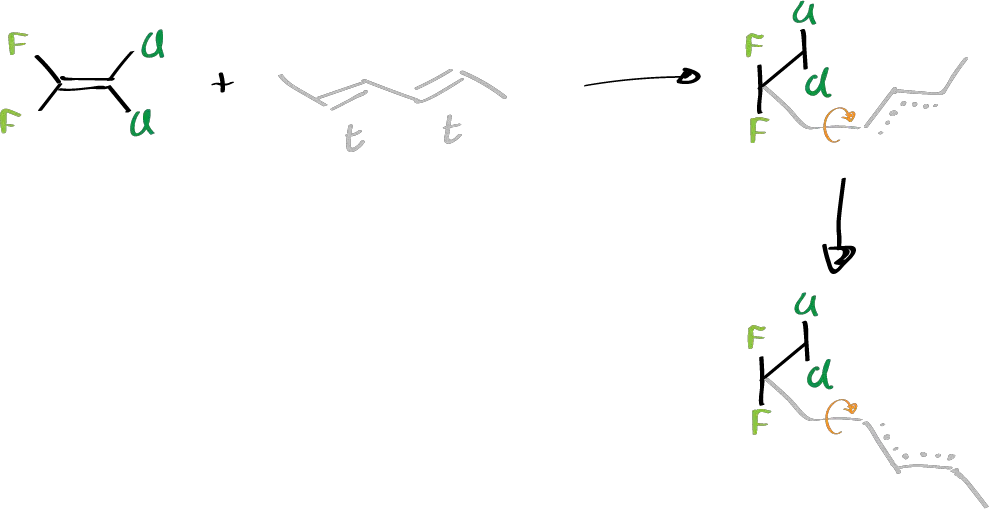
Therefore, it can now cycle in one position or the other. If it cycles in the first it will give rise to the first product of the reaction, whereas if it cycles the second it will give rise to the second product, but both have the trans configuration in the double bond because of the existence of that allyl radical, and of the two possibilities of regiochemical attack you get only the one in which the position of the fluorine atoms is the one in the reaction. This is because a free radical is not stable in the vicinity of two fluorine atoms.
However, if it is stable in the vicinity of two chlorine atoms, the attack is preferably obtained first to give rise to the radical attached to the carbon having the two chlorines which can evolve and cycle to give rise to the two reaction products.
Several experimental studies have been attempted to try to find 2+2 cycloaddition reactions, even if only by looking for the possibility that one of the components acted antarafacially to see if a 2+2 reaction can indeed occur.
For an antarfacial interaction to occur in a two-electron component, an approach geometry has to be produced as shown:
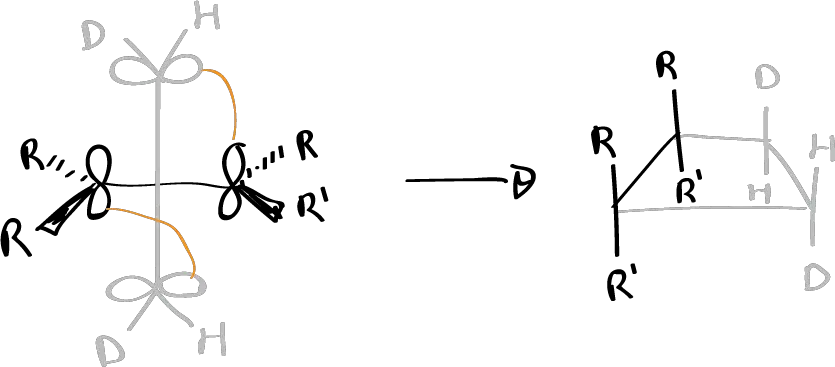
For this to occur we have to have the substituents on each atom in such a way that the substituents on the olefinic system that has the π orbitals in the perpendicular plane place those substituents on the plane of the paper. And the olefinic system that is approaching vertically must have the substituents in the plane perpendicular to the plane of the paper, so that one of the substituents goes inward and one outward.
For a reaction of this type to be studied experimentally, the substituents must be suitable so that a certain stereochemistry can be observed.
Therefore, in order to demonstrate the existence of an antarafacial interaction, there must be a certain stereochemistry in the reactants, which must be conserved in one of the components and there must be an inversion of the configuration in the one acting antarafacially, and also, since one must act in a perpendicular manner and bringing the groups closer to the other, those groups that come closer must be as small as possible so that there is the minimum possible steric interaction.
This has been attempted by reacting these two compounds.
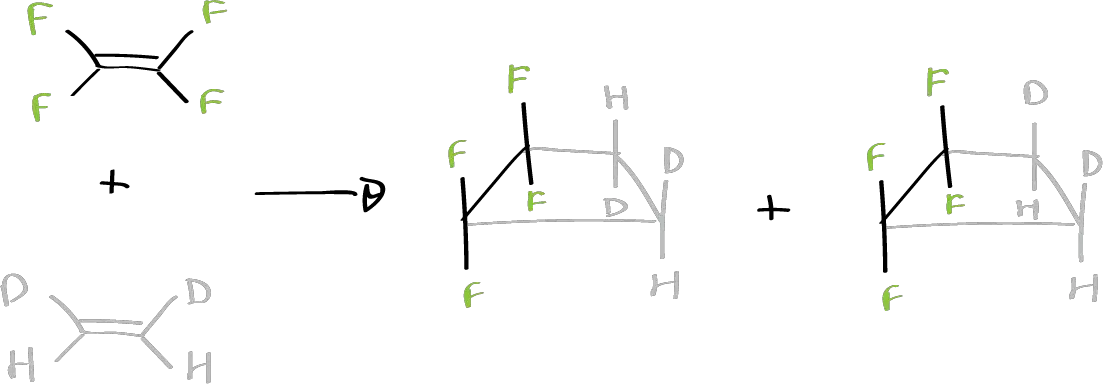
This is the possible reaction, in which the sterochemistry of the antarfacially acting component can be observed with the smaller substituents approaching. So, when stopping the cis or trans isomer, in both cases the same mixture of products is obtained in which the configuration is distributed. That is, when there are 4 substituents in the two olefinic systems involved, the reaction proceeds by a radical mechanism since mixtures of products are obtained. A supra-supra reaction does not take place since it is forbidden by symmetry but the supra–antara is forbidden by steric interactions or by the tension involved in the twisting of one of the components.
Seeing that the antarafacial reaction could not be observed because there is always some interaction even if we put little bulky substituents. You can think of putting a reagent that does not have these substituents, this would be the case of ketenes, where there are no substituents on oxygen and if on carbon and we can try to observe the reactions of these ketenes with properly substituted olefins to see if not having one of the substituent atoms, if you can give the reaction antarafacially.
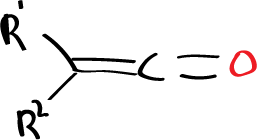
Very interesting experimental features have been obtained which suggest that the reaction is in fact a pericyclic reaction in which one of the components is attacked in an antarfacial way.
When substituted ketenes (with different substituents: one large G and one small P) are reacted with an olefin also with large and small substituents, the attack of the ketene is obtained in a certain way, which is the way that gives rise to the 4-membered cyclic system in which the large substituents are on one side and the small ones on the other.
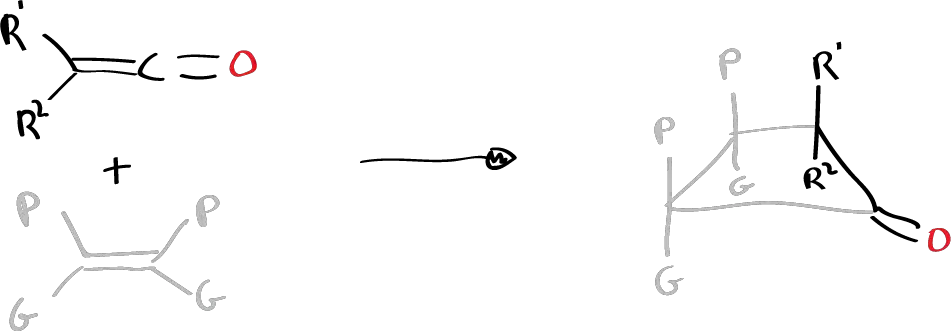
Also when suitably substituted ketenes are reacted with cyclopentadiene in a 2+2 reaction, only the endo product is obtained with respect to the small group R2.
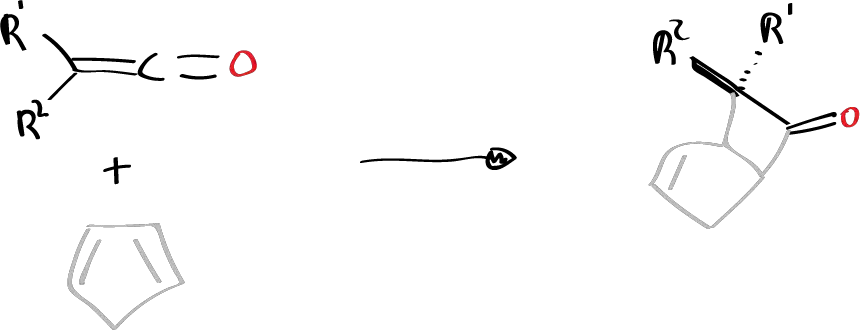
These peculiar characteristics could be due to the fact that the ketene reacts in an antarfacial way which explains the stereochemistry of the reaction. The olefin and ketene will be oriented as indicated.

Of the two possibilities of approaching the ketene, which can be with the carbonyl group upwards or downwards, the one in which less steric interactions are established will be the one in which the substituents will go downwards and as they are larger than the carbonyl, it will be possible to establish a steric interaction with the large groups L, so that the carbonyl, being the smallest part S of the ketene, will be placed with the orientation towards the large groups L.
Of the two groups that the ketene has, the small one will approach the ethylene molecule, as we have drawn it. This must be the geometry of approach to the TS for an antarfacial interaction to occur on the part of the ketene. Once it cycles it evolves into a product where the small groups are all on the same side.
This has been seen with examples so that if you stop from this ketene and react it with cis-butene you get exclusively the product that has the bulky group downwards and the cis-butene sterochemistry is maintained. Thus, it can be said that this is a pericyclic reaction in which one of the components acts in an antaraphase-like manner.
Five-membered ring formation
We are looking at cycloaddition reactions from an experimental point of view, we are trying to see examples where it can be shown that there is a pericyclic reaction or other cases where it can be thought that there is a certain pericyclic mechanism, to show that it is not so (they go against the generalized Woodward-Hoffmann rules).
There is a whole series of cycloadditions that allow the formation of 5-membered rings, for example those known as 1,3-dipolar cycloadditions, which are π2+ π4 reactions and therefore must be allowed by symmetry.
There are a large number of 1,3-dipole reactions (dialkenes) that react very easily to give rise to different types of heterocyclic systems. It is one of the reactions that show the greatest synthetic potential for synthesizing heterocycles.
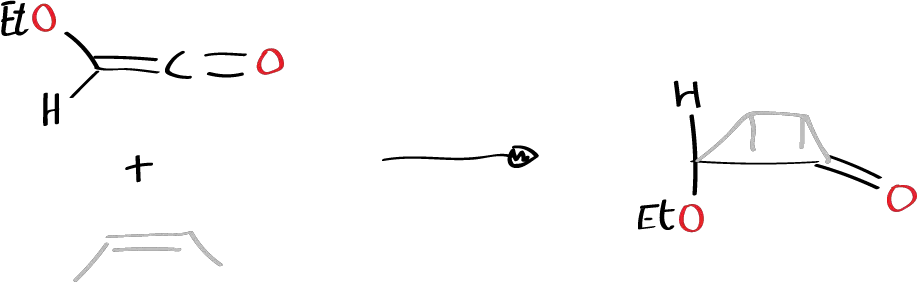
These reactions are stereospecific and the idea is that they are pericyclic reactions, which does not mean that the TS has to be totally symmetrical, that all the bonds that have to be formed are equally formed and that those that have to be broken are broken. There may be an TS in which a certain charge is more displaced in one of the regions than in another and one of the bonds is more formed than another. But this does not detract from the fact that the reaction is concerted and takes place in a single step. Normally, many examples of cycloaddition reactions involving some 1,3-dipole of the many that can occur are known, such as nitrous oxide, nitrile oxides, ozone, carbonyl oxide. All these compounds have structures of what are known as 1,3-dipole and are characterized by having 4 p electrons in a system of 3 atoms, and can give rise to cycloaddition reactions with olefins and will generate a 5-membered ring. Thus, for example, when such a structure is reacted with a double bond system:
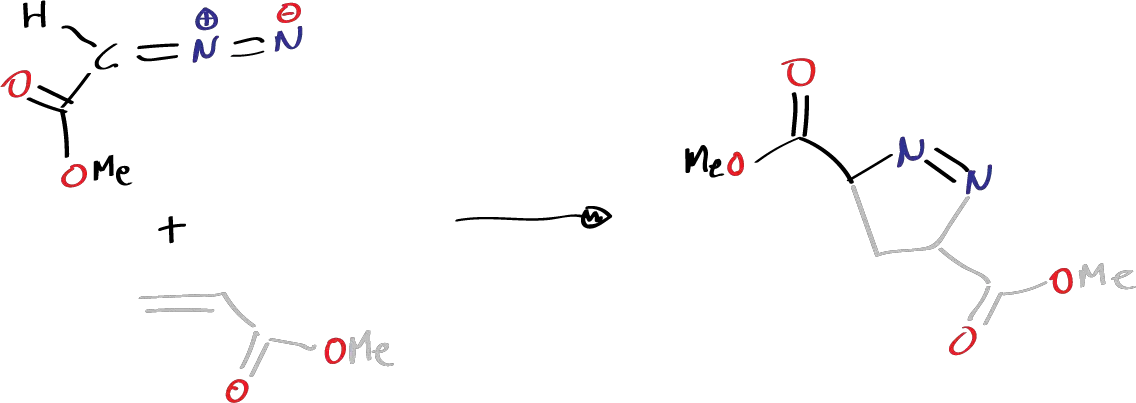
These reactions occur very easily at low temperatures (in carbonic snow) and give rise to the corresponding heterocycles.
Other cycloaddition reactions are, for example, ozonolysis of alkenes. Alkenes react with ozone also very easily at low temperature (carbonic snow or even less) so that the accepted mechanism is one that features a series of cycloaddition reactions:
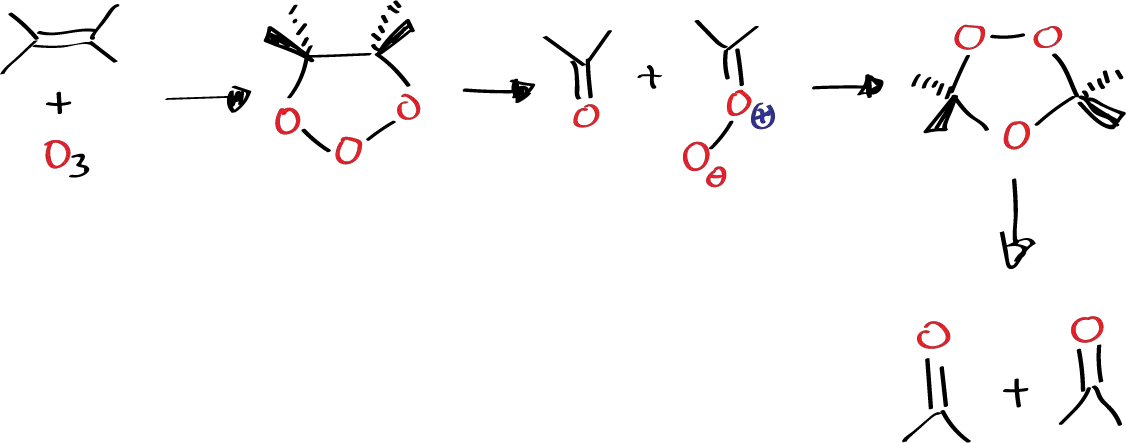
In a first stage there would be a cycloaddition of the olefin to ozone to give rise to what is known as primary ozonide, which is a very unstable structure that produces a cycloreversion, but instead of going backwards to give ozone again, it gives rise to a carbonyl and carbonyl oxide which is also a 1,3-dipole structure. So, if we have a carbonyl oxide which is a 1,3-dipole and a carbonyl which is a two-electron p-system another cycloaddition can occur, when the carbonyl system has a suitable structure and is not too impeded. In this case the cycloaddition would give rise to what is known as ozonide, which are already more stable structures and can even be characterized. As they have the peroxo group, they are potentially dangerous structures and what is usually done is to destroy them to give rise to two products that may be the same or different depending on whether the olefin was symmetrical or not. If the breakage is done in an oxidizing medium, each carbon passes to its maximum oxidation state whenever possible, i.e. to carboxylic acid or to ketone if further oxidation is not possible.
There is a great deal of evidence to suggest that it is indeed that mechanism that occurs, the Creege mechanism.
It has been possible to detect the primary ozonide when the ozonolysis is performed at very low temperature, below -100ºC, and the reaction product is studied by NMR at the moment it is produced. This is done by performing the reaction in a tube inside the NMR apparatus. Under these conditions, by recording the spectrum before adding ozone and then recording spectra every few seconds and increasing the temperature slowly, the evolution of the products can be seen. When the reaction is done with this alkene:
The peak corresponding to the reactant can be easily recognized from the peak corresponding to the first product of the reaction which is the primary ozonide and that evolves later to the formation of the secondary ozonide.
There are other tests that allow us, for example, to detect carbonyl oxide, which is also a very unstable structure that cannot be isolated, but can be generated in situ by procedures other than ozonolysis. Or they can be trapped by something other than the carbonyl resulting from the reaction, to try to demonstrate its existence.
The carbonyl oxide can be generated instead of by ozonolysis, by photolysis reactions. When this carbonyl oxide is generated in the presence of aldehydes, for example in the presence of acetaldehyde, the reaction takes place in such a way as to give rise to the ozonide corresponding to the reaction of that carbonyl oxide with that aldehyde.
So we can say that carbonyl oxides if formed react with aldehydes to give rise to oxonides. This is not a necessary way of implying that the carbonyl oxide is an intermediate in the reaction but if formed the carbonyl oxide reacts with the aldehydes.
Another way to detect carbonyl oxide is to start from an olefin structure in which the carbonyl oxide formed cannot react with the carbonyl also formed in the reaction. For example, in a tetrasubstituted olefin a primary ozonide will be formed which will evolve towards cleavage to form acetone and the corresponding carbonyl oxide.
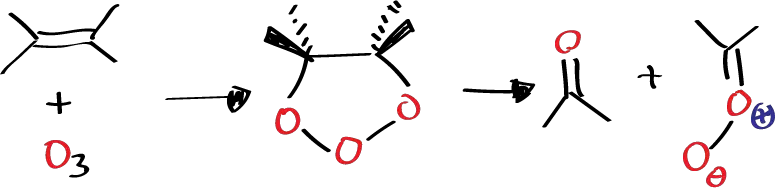
If the reaction is carried out in an aprotic solvent such as methylene chloride, this carbonyl oxide is unable to react with the ketone due to steric hindrance, so under these conditions it reacts with itself, dimerizes to give rise to this ozonide which is a paradoxically absolutely stable product that can be crystallized, its melting point can be calculated, etc.

That is, when we do an ozonolysis reaction with a structure in which the second cycloaddition reaction cannot occur to give rise to the ozonide, then the carbonyl oxide, if it cannot react with anything else, dimerizes giving rise to structures that are stable. The reason why 3,3,6,6-tetramethyl-1,2,4,5-tetraoxane (diacetone peroxide) are so stable having as they do two peroxo groups may be due to the stability of the 6-membered cyclic system since what we have is a conformation in which all the angles are tetrahedral and dihedral angles have an alternating structure, etc.

The reaction of 2,3-dimethylbutene with ozone, instead of being done in dichloromethane it is done in ethanol, the carbonyl oxide also cannot react with the ketone that is formed, but now it can react with the solvent, so that this structure would result.

This hydroperoxide is somewhat unstable but can be characterized in a way that demonstrates somewhat that carbonyl oxide is formed. Since both this hydroperoxide with the above dimer can only come from the reaction of carbonyl oxide.
If you do the same reaction in the presence of an aldehyde, you get the ozonide resulting from the reaction of the carbonyl oxide formed with the aldehyde we have put in.
All this is more than conclusive evidence to think that this is indeed the mechanism that occurs in a normal ozonolysis in which the olefins are either not very impeded or are not very rich in electrons. As we can see, it is a series of two cycloadditions and a cycloreversion.
In general there is much experimental evidence to suggest that 1,3-dipole reactions are pericyclic reactions. But as always alternative mechanisms are proposed. Theoretically it cannot be a radical mechanism but it can be a dipolar mechanism, since the 1,3-dipoles are structures that have an important charge separation and can produce a mechanism in which a two-step attack takes place. A first attack of the area of excess positive charge of the olefin giving rise to another dipole that can subsequently close.
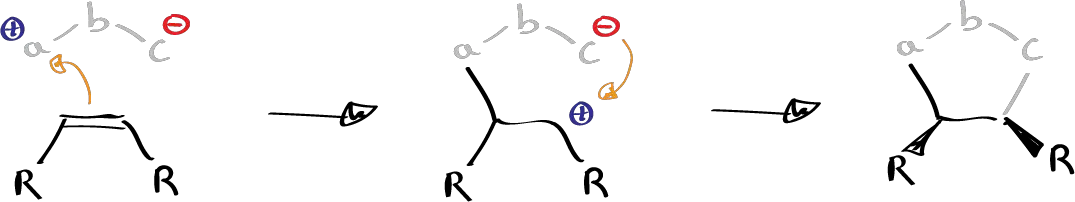
If that cycles by electrostatic interaction in a very fast way before the twist around that bond can occur, it will give rise to the cycloaddition product in which the stereochemistry of the originating olefin is maintained.
If, on the other hand, this reaction is not so fast and allows time for rotation to occur around the bond, then we would have a mixture of the two stereoisomers.
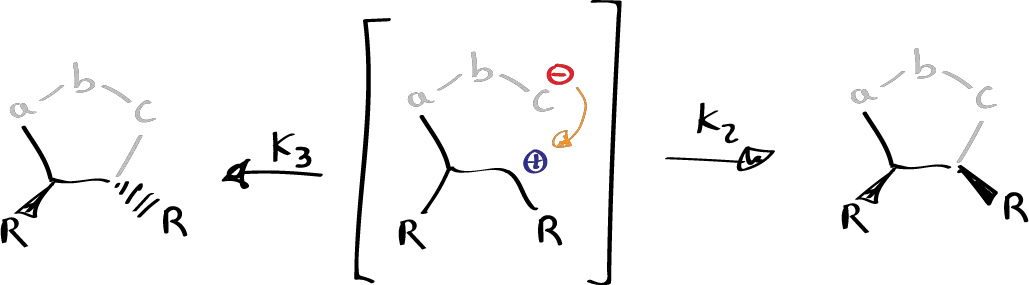
Firestone proposed this type of mechanism and said that it is not that a pericyclic reaction occurs but that k2 is much faster than the k3 that gives rise to the spin.
It is possible that depending on certain variables one mechanism or another may occur. But in general it can be said that 1,3-dipolar cycloaddition reactions are usually pericyclic reactions, in some special cases where there are very important steric effects it may be the case that they are two-step reactions where a dipolar attack occurs where an intermediate is produced in which one bond has been formed and the other is still unformed.
Full Professor of Organic Chemistry at the University of Granada, with a long-standing research career in Computational Chemistry and molecular modeling and design.