
What are three-membered heterocycles?
The chemistry of three-membered heterocycles is dominated by angular tension, which leads to high reactivity.
Attack by a nucleophile on a ring carbon, heating or irradiation can result in ring opening.
Therefore, three-membered heterocycles are abnormally reactive molecules, and very useful as intermediates in synthesis.
Oxiranes (epoxides)
Oxiranes or epoxides are very common reagents in organic chemistry, as they can easily and highly selectively form or destroy the ring system.
They are present in many compounds of biological interest. Aromatic hydrocarbon-derived oxiranes (arene oxides) are intermediates in aromatic hydroxylation reactions in living systems.
Oxiranes derived from simple alkenes, by catalytic reaction with oxygen, are large-scale industrial chemicals used for the manufacture of polymers.
There are different procedures for the synthesis of oxiranes, almost all of them starting from olefins.
Synthesis of oxiranes by epoxidation of olefins
The most common reagents used for this reaction are perocarboxylic acids (peracids).
When an alkene is reacted with an organic peracid, the π bond is broken, forming a 3-membered cyclic ether.
The most commonly used peracids are perbenzoic acid, monoperphthalic acid and peracetic acid.
The peracids are electrophilic and attack the π-cloud of the alkene. This attack is favored if electron releasing substituents are present.
The mechanism for the attack is believed to be as follows:
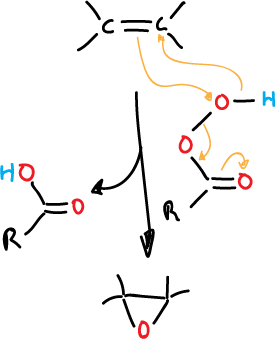
The reaction is stereospecific, proceeding via cis-addition, as illustrated in the figure below:
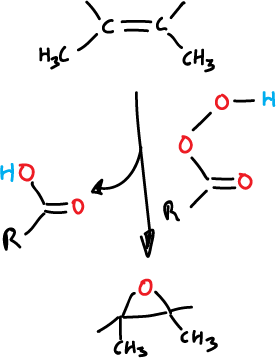
Epoxidation is performed on the less steric hindered side of the olefin.
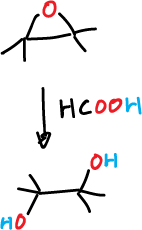
Perfformic acid cannot be used, since the released formic acid is very strong and would bind the epoxide forming a transdiol.
When the olefinic bond is conjugated with a group that attracts electrons strongly, such as the carbonyl (>C=O) or cyano (—C≡N) group, the rate of epoxidation slows down, or the reaction is no longer affected.
Epoxidation with alkaline hydrogen peroxide (HOOH) (HO⊖) is commonly used for systems of this type.
The reaction proceeds by Michael addition of the hydroperoxide anion to the unsaturated system, followed by intramolecular displacement of the hydroxyl ion.
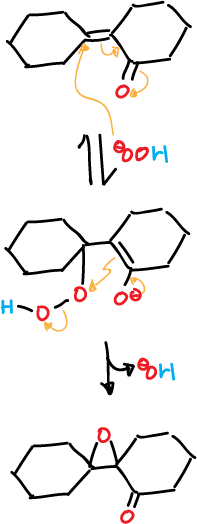
Epoxidation with hydrogen peroxide is not stereospecific, as in the case of peracid. However, a single epoxide is usually formed, which is not stereochemically related to the reagent.
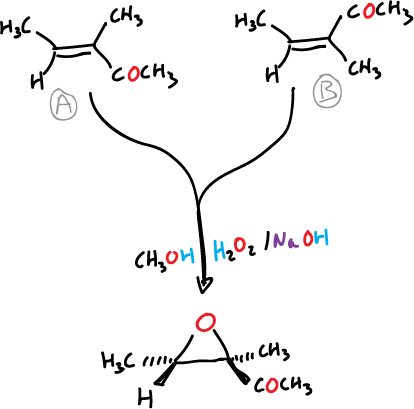
For example, oxidation with peroxide of the isomeric ketones A and B produces the same epoxide, as shown in the scheme.
The α,β-unsaturated nitriles, under these conditions produce α,β-epoxyamides.
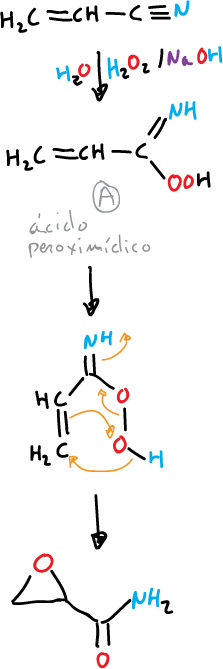
The reaction is carried out by the intermediate A (peroxyimidic acid) which functions as an electrophilic reagent.
Synthesis of oxiranes from halohydrins
In fact, this method also starts from olefins since halohydrins are derived from olefins.
Darzens reaction of oxirane synthesis
The Darzens reaction starts from carbonyl compounds. It can be represented by the addition of ethyl chloroacetate to the carbonyl group of acetophenone, under conditions of catalysis of a very strong base such as potassium terbutoxide(tBuOK) or sodium amide (NaNH2).
Initially, a halohydrin (chlorohydrin) is formed, which in a basic medium gives rise to the epoxide.
fig-09
The reaction is reversible until it anhydrifies, then it becomes irreversible.
The Darzens reaction does not usually end in the epoxide, but with a base such as sodium hydroxide (NaOH) gives rise to an aldehyde, resulting in dexcarboxylation (loss of CO2).
fig-10
Synthesis of oxiranes by elimination of a tosylate
This reaction starts from an alcohol, and is related to the elimination of HX in halohydrins.
fig-11
Synthesis of oxiranes by the reaction of aromatic aldehydes with phosphoric triamides.
Triamin phosphines react with aromatic aldehydes to give oxiranes, as shown in the figure below:
fig-12
The proposed mechanism consists of the following steps.
fig-13
Industrial production of oxiranes by oxidation of olefins
This procedure is good for obtaining ethylene oxide, but it causes problems with propylene, which tends to oxidize the methyl in the allylic position. It is carried out with hydroperoxides, e.g. ethylbenzene hydroperoxide or tertiary butyl hydroperoxide (tButOOH).
fig-14
The reaction is carried out in two steps:
- In a first step, oxygen and olefin are mixed to give the corresponding hydroperoxide.
fig-15
- In a second step, the hydroperoxide oxidizes the olefin, forming the corresponding alcohol.
fig-16
Reactivity of oxiranes
The marked reactivity of oxiranes is due to the ring tension. The average bond angles of 60° of a triangle are considerably smaller than the tetrahedral angle of carbons (109.5°), or that of divalent oxygen (110°).
In the following, the reactions that take place in acidic and basic media are listed first.
Acid-catalyzed oxirane opening
In general, epoxides or oxiranes can be protonated like ethers, but once protonated they can undergo attack by various nucleophiles, resulting in anti-hydroxylation.
fig-17
When the epoxide is asymmetric the attack of the nucleophile occurs in the position shown in the figure below:
fig-18
If the reaction is carried out with gaseous hydrogen chloride (HCl) or hydrogen bromide (HBr), the reverse reaction to the synthesis of epoxides from halohydrin occurs.
fig-19
Ethylene oxide can polymerize, in acid medium as shown in the scheme:
fig-20
Base-catalyzed oxirane opening
Oxiranes are further differentiated from ordinary ethers by this type of ring opening. Thus, the attack is suffered by the epoxide itself, not by the protonated epoxide.
fig-21
When this reaction is carried out in an acid medium, it proceeds as shown in the following scheme, giving the same product as in a basic medium.
fig-22
Although the above two reactions give the same product, there are differences between carrying out the reaction in an acidic or basic medium. This is evident when an asymmetric epoxide is used as starting material.
fig-23
In acidic media, the nucleophile’s preferred site is the central carbon, because the charge is best accommodated in that position.
fig-24
On the other hand, the attack of the nucleophile in basic media takes place on the less sterically hindered carbon.
fig-25
Summarizing the two types of reactions:
- In basic media, steric interaction predominates.
- In acidic media, electronic interaction predominates.
Opening of oxiranes by addition of other nucleophiles
Other examples of addition of other nucleophiles of different types (NH3, SH2, CH3MgBr, AlLiH4), which also produce oxirane opening, are shown in the following schemes.
fig-26
Transposition of oxiranes to dihydrofurans
Vinyloxiranes can undergo transposition to dihydrofurans by a reaction involving 1,5-dipolar cyclization.
fig-27
Oxirane deoxygenation
Epoxides can be easily deoxygenated by tertiary phosphines at elevated temperatures (150-200 ºC) producing olefins.
fig-28
Depending on the configuration of the epoxide the mixture of alkenes would be mostly in the alkenes (Z or E), as shown in the scheme. The cis epoxides give mostly E alkenes, and the trans ones give rise to Z alkenes.
The reaction is carried out by nucleophilic attack of the tertiary phosphine on a carbon atom of the epoxide, yielding a product similar to betaine.
fig-29
This, when rotated 180º at the central C—C bond, breaks and releases the phosphine oxide to give as a predominant product an olefin of the opposite configuration to that of the initial epoxide.
Examples of the use of oxiranes in synthesis
- The Darzens reaction for the formation of glycidic acid, encompasses the reaction or condensation of a ketone or aromatic aldehyde with an α-haloester, in the presence of a strong base such as potassium terbutoxide (tBuOK) or sodium amide (NaNH2).
The product, initially formed (aldol condensation product), is closed to an oxirane ring intramolecularly. These substances are called glycidyl esters or α,β-epoxyesters.
- Epoxides can be used as alkylating agents for methylene-active compounds.
fig-30
- The reaction of epoxides with dimethyl sulfoxide (DMSO) generates dihydroxyalkyl dimethylsulfonium saltswhich, in basic media, form the corresponding acyloins (α-hydroxyketones).
fig-31
Aziridines
Like the oxiranes, aziridines are also very labile heterocycles. They have a high tendency to open, making them good alkylating agents. For this same reason, many of the aziridines are toxic and mutagenic agents.
They exist in nature, for example, the antibiotic and antitumor mitomycin C, whose antibiotic character is associated with the aziridinic ring.
fig-32
Ring synthesis
Almost all aziridine ring synthesis reactions are closure reactions caused by intramolecular substitutions. These closures are very similar to those employed in the synthesis of oxiranes.
The starting products are usually amino alcohols, which in turn can be obtained from oxiranes by reaction with ammonia(NH3). Normally the hydroxyl group (–OH) is transformed into a good leaving group with the appropriate reagent (sulfonate or benzene sulfonate group).
Wenker aziridine synthesis
Wenker synthesis is the conversion (in acidic medium) of a β-amino alcohol to a β-aminohydrogen sulfate that cyclizes by treatment with a strong base.
fig-33
Obtaining α-haloamines and subsequent cyclization.
Another method of obtaining aziridines would be the conversion of the amino alcohol into β-haloamine and its subsequent cyclization as shown in the figure.
fig-34
The α-haloamines can be obtained from alkenes as follows:
fig-35
From olefins and iodine isocyanates
Aziridines can also be synthesized from olefins if treated with iodine isocyanate.
fig-36
Under basic conditions, the NH of the carbamate group is partially deprotonated. Thus, the resulting anion achieves an SN2-type internal displacement, leading to the cyclized carbamate which in turn hydrolyzes and generates the corresponding aziridine.
fig-37
1,3-Dipolar cycloaddition between azide and olefins
Another synthetic route for the preparation of aziridines consists of the 1,3-dipolar cycloaddition reaction of azides to olefins, as indicated in the following scheme.
fig-38
Inermediate 1,2,3-triazoles with retention of the starting olefin configuration are formed which either thermally or photochemically generate the corresponding aziridinine with loss of nitrogen (N2).
Nitrogen functionalization
The nitrogen of aziridines (N—H), or the nitrogens of N-alkylated aziridines, is nucleophilic. Therefore, it can behave as secondary or tertiary amines. Thus, it is susceptible to alkylation with haloalkanes, epoxides and similar reagents. In this way, they undergo conjugate addition reactions that generate nitriles and a,b-unsaturated carbonyl compounds.
On the other hand, N-chloro and N-acilaziridines can also be prepared from compounds without substituents on the nitrogen.
Some examples of where replacement products can be isolated with good performance are given below:
fig-39
Ring-opening reactions
The ease of ring opening by nucleophiles depends on the electron-accepting properties of the N—R(H) substituent, steric effects on the ring carbons and the nature of the attacker. Thus, the breakage takes place on the least substituted carbon.
fig-40
Fragmentation reactions
Nitrogen unsubstituted aziridines are stereoselectively deaminated with nitrosyl chloride (NOCl). The mechanism is probably from N-nitrosoaziridine.
fig-41
Thiiranes (ethylene sulfide)
The thiiranes, or ethylene sulfides, have much in common with the other 3-membered heterocycles. However, part of their chemistry is specifically associated with the presence of the sulfur atom.
For example, there are two oxidized versions of the ring system, the monoxide and the dioxide shown in the schematic.
fig-42
Synthesis of thiiranes
They are obtained from oxiranes, by reaction with thiocyanate ions or thiourea.
fig-43
Reactivity of thiiranes
Analogous to aziridines and oxiranes, thiiranes undergo annular cleavage by the action of dilute acid or base.
fig-44
2H-Aziridines
Of the two possible azirin isomers, only 2H-azirin has been isolated and characterized. However, it is unstable above the temperature of liquid nitrogen (-196 °C).
fig-45
Many other aziridines are low melting point isolable liquids or solids. In addition, most of the known compounds of this type have an alkyl, aryl or dialkylamino substituent at the 3-position.
fig-46
Synthesis of 2H-aziridines
The most general method of synthesis of these compounds is the thermal or photochemical decomposition of vinyl azides. This reaction can occur via vinylnitrenes intermediates.
fig-47
Reactivity of the 2H-aziridines
Ring strain plays a notable role in determining the chemistry of these compounds. Three types of reactions are possible:
- Reactions at the C3=N1 bond.
- Rotura térmica del enlace N1—C2.
- Breakage of the C2—C3 bond with ultraviolet light (photochemically).
Reactions at the C3=N1 bond of 2H-aziridines
This bond is susceptible to nucleophilic attack.
The lithium aluminum hydride (LiAlH4) reduces the bond with formation of aziridines. Moreover, the reduction is stereoselective and takes place on the less impeded side.
A similar reaction occurs with Grignard reagents.
fig-48
Thermal cleavage of the N1—C2 bond of 2H-aziridines
This thermal cleavage reaction of the N1—C2 bond is the reverse of the vinylnitrene cyclization.
Thermolysis often results in the breaking of this bond. However, by-products of breaking the C—C bond are sometimes found.
When azirines, with conjugative substituents at C2, are heated, the main product is often a 5-membered ring. This ring is a derivative of a transposition that is formed by bond cleavage and recycling.
fig-49
Photochemical C2—C3 bond breaking
Ultraviolet light breakage (photolysis) of aziridines often involves C—C bond breakage and formation of nitrile ilydes.
fig-50
Three-membered heterocycles with 2 heteroatoms
fig-51
They can be synthesized relatively easily but their reactivity is very high and they possess certain unusual properties.
Synthesis of oxaziranes
The preparation of these compounds is easily achieved by direct insertion of a suitable heteroatom into a carbonyl or imine double bond.
- Oxaziranes are synthesized by oxidation of imines with organic peracids. It is a different reaction from olefin epoxidation, which was stepwise and protonated imine intermediates were formed. In contrast, in imine oxidation the by-producs formed are nitrones.
fig-52
Another method of synthesis would be the reaction of aldehydes and ketones with chloroamines (CH3NHCl).
fig-53
The reaction mechanism involves an initial 1,2 addition of the nitrogen-bearing component to the carbonyl group. This process is followed by an intramolecular SN2 shift.
fig-54
Synthesis of diaziridines
Diaziridines can be prepared from ketones by reaction with ammonia, or a primary amine, together with an aminating agent such as chloramine or hydroxyl-amino-O-sulfonic acid.
fig-55
Another method of electrocyclic closure of stabilized azomethinimides occurs in the presence of ultraviolet light, and is reversible.
fig-56
In addition, other diaziridines with electron-attracting (conjugative) substituents also break down with heat.
Synthesis of 3H-diazirines
3H-diazirines are cyclic isomers of diazoalkanes and in fact some diazirines have been obtained by photoisomerization of diazoalkanes. However, the easiest way to obtain them is the oxidation of the unsubstituted diaziridines with: silver oxide, potassium permanganate, mercuric oxide, etc.
Reactivity of oxaziranes
Oxaziranes are active oxygenated compounds, largely comparable to organic peroxides. The oxazirane ring usually decomposes slowly in the presence of strong acids, but is stable to weak basic reagents, although there are exceptions.
Stability varies depending on the nature and number of substituents in the molecule.
Normally, the oxazirane ring is broken in all its reactions. As, for example, the formation of imines and amines in the following scheme.
fig-57
Acid hydrolysis occurs with formation of an aromatic aldehyde and an alkylhydroxylamine.
fig-58
This reaction shows configurational stability in nitrogen.
Also, N–sulfonyloxaziridines are excellent oxygen transfer reagents.
For example, the optically active compound, camphorsulfonoyloxaziridine, and its enantiomer have been used as asymmetric oxygen transfer reagents.
fig-59
Although the oxazirane ring is stable against basic reagents, however, oxaziranes with 2-methylene or 2-methinyl substituents react vigorously with alkali in aqueous or alcoholic solution. They produce ammonia in quantitative form.
fig-60
Reactivity of diaziridines
Diaziridines, which are also a type of oxidizing agent, are more stable than oxaziranes. In addition, they are slightly basic and form salts but also hydrolyze easily in acid solution to give a carbonyl compound and a hydrazine. On the other hand, they are stable against alkali.
They can be catalytically hydrogenated and give two amines, but with lithium aluminum hydride (LiAlH4) other types of amines are obtained.
fig-61
In acid hydrolysis, the increase of substituents on the carbon atom accelerates hydrolysis.
fig-62
On the other hand, diaziridines, which contain at least one NH group, undergo certain reactions characteristic of secondary amines, with retention of the ring.
For example, the condensation of diaziridines with chloral (trichloroacetaldehyde).
fig-63
Reactivity of 3H-diazirines
The diazoalkane isomers have notable differences with the 3H-diazirines, such as being more stable and less reactive, except for their explosive nature (in the low molecular weight derivatives). However, acids and alkali do not react with the 3H-diazirine ring at room temperature.
Thermal or photochemical decomposition of 3H-diazirines results in the release of nitrogen, in the presence of nitrobenzene, as illustrated in the following scheme.
Photolysis in the presence of trans-2-butene produces mainly trans-cyclopropane.
fig-64
Also, they can undergo reactions with ring retention. For example, the addition of Grignard reagents.
fig-65
Full Professor of Organic Chemistry at the University of Granada, with a long-standing research career in Computational Chemistry and molecular modeling and design.