
Oxidation
Oxidation of aldehydes
Aldehydes are more easily oxidized than ketones. In the presence of strong oxidizing agents such as KMnO4 or hot HNO3, they lose the hydrogen bonded to the carbonyl and give the corresponding acid. One of the most commonly used oxidizing agents is Jones’ reagent (CrO3 in aqueus H2SO4), which has the disadvantage of requiring acidic conditions and can also react with other functional groups present in the molecule.
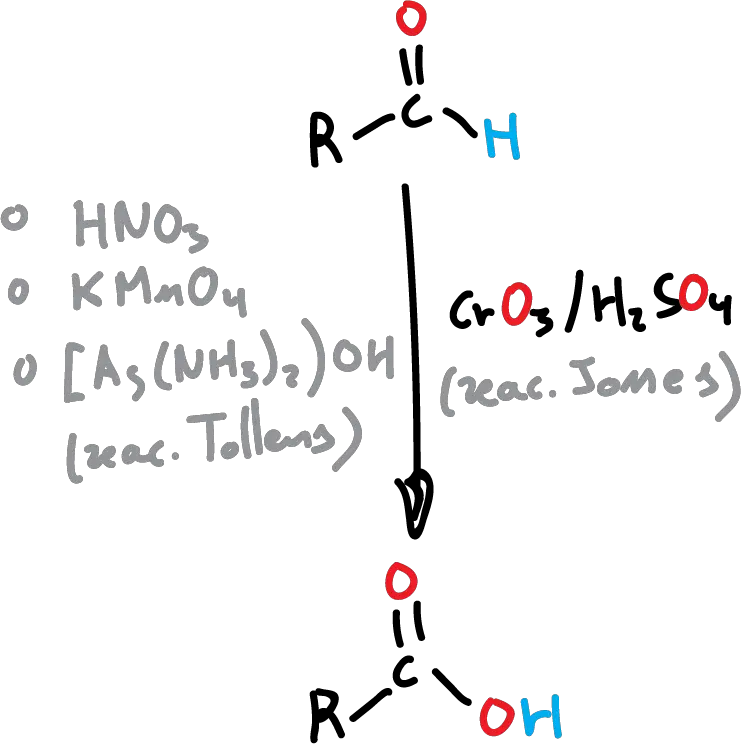
In these cases, and for aldehydes, the Tollens’ reagent (Ag2O in aqueous NH3 ) is commonly used, with the advantage that the reaction is chemoselective (only this function is oxidized in the molecule). In contrast to aldehydes, ketones only react with strong oxidants by breaking the C-C bonds to give mixtures of carboxylic acids.
Baeyer-Villiger oxidation
Ketones can be oxidized to esters in the presence of carboxylic peracids (R’CO3H)). Among the most commonly used carboxylic peracids is meta-chloroperbenzoic acid (MCPBA).
Non-symmetric ketones produce mixtures of esters, since oxygen can be inserted on both sides of the carbonyl carbon. To predict the majority compound formed, the migration aptitude of the substituents in the Baeyer-Villiger reaction must be known:
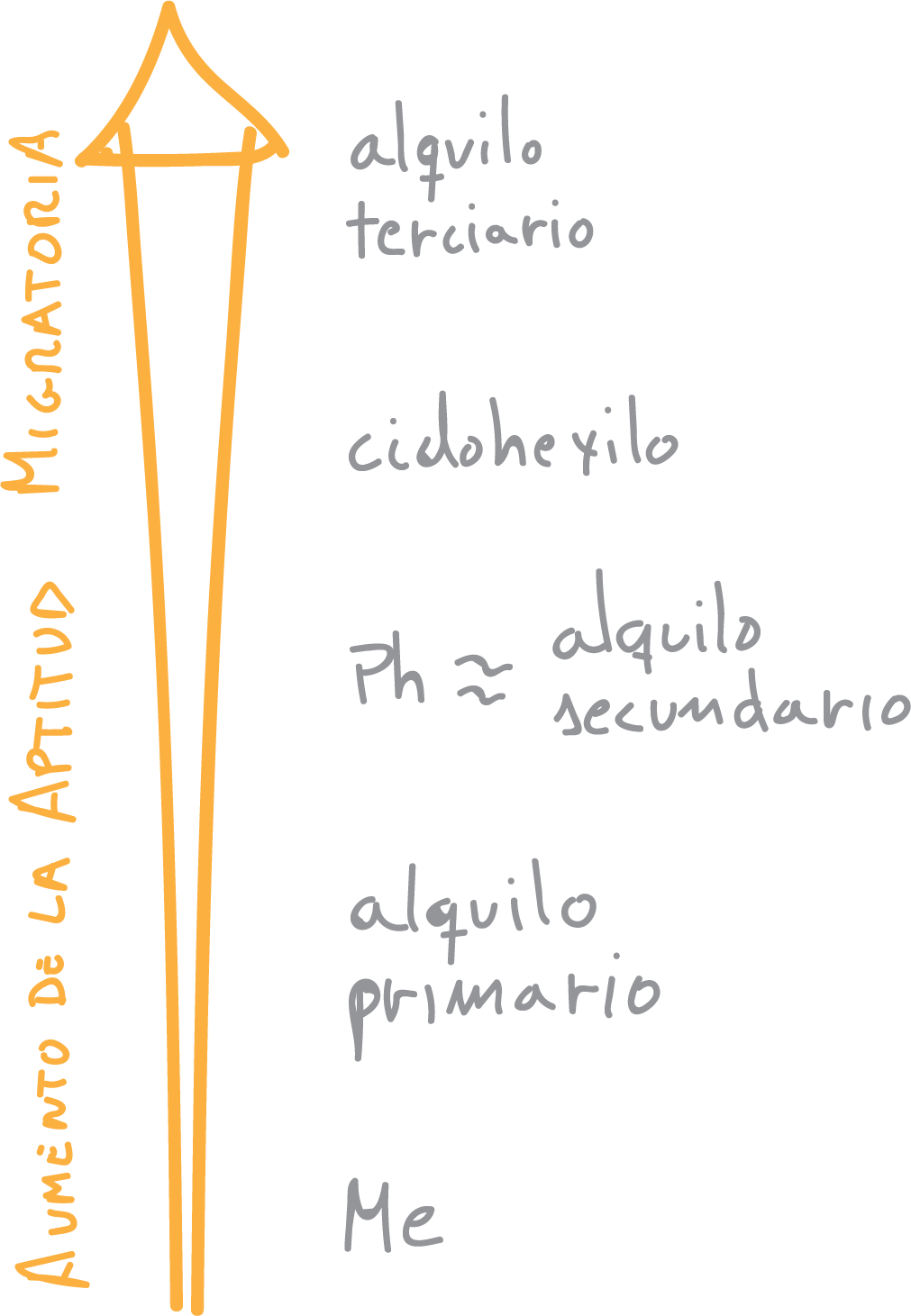
It should be noted that some groups exhibit charge stabilization/destabilization effects of the transition state, as well as steric effects that may alter this order of priority.
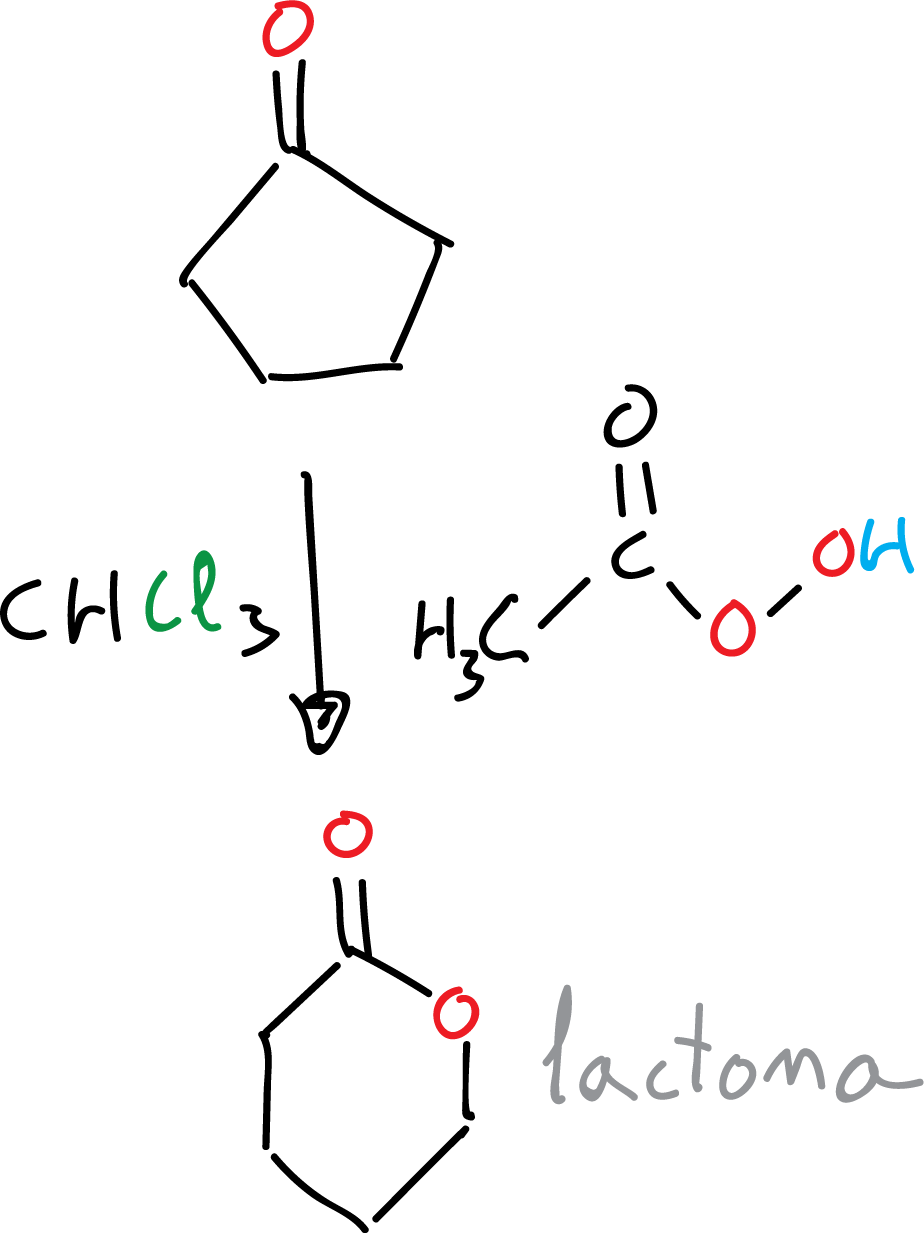
In general, the tendency to migrate improves with increasing substitution. On the other hand, cyclic ketones give rise to cyclic esters, commonly called lactones:
Reduction
Clemmensen reduction to alkane
It is carried out under acidic conditions (HCl) with zinc amalgam, Zn(Hg), which allows aldehydes and ketones to be reduced to alkanes.
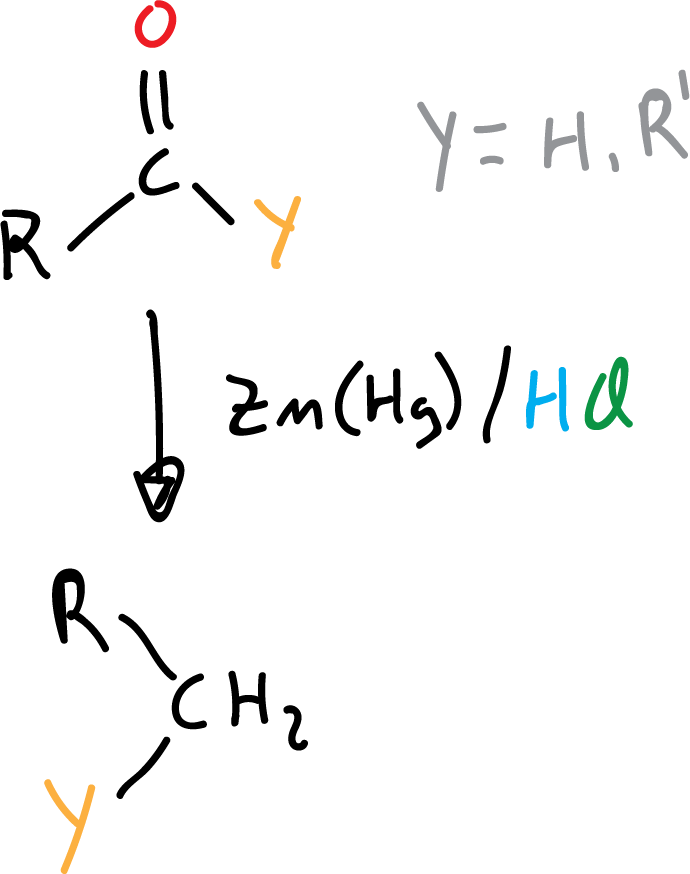
The starting aldehyde or ketone must be stable in an acidic medium, otherwise we must use the Wolff-Kishner reduction that takes place in a basic medium.
Wolff-Kishner reduction to alkane
The Wolff-Kishner reaction, along with the Clemmensen reaction, reduce the >C=O carbonyl group to methylene >CH2. Both reactions do not reduce the functional groups >C=C<, -C≡C- and -COOH, which may be present in the molecule. The choice of one or the other reduction reaction is given depending on the sensitivity of other functional groups present in the molecule under acidic or basic conditions.
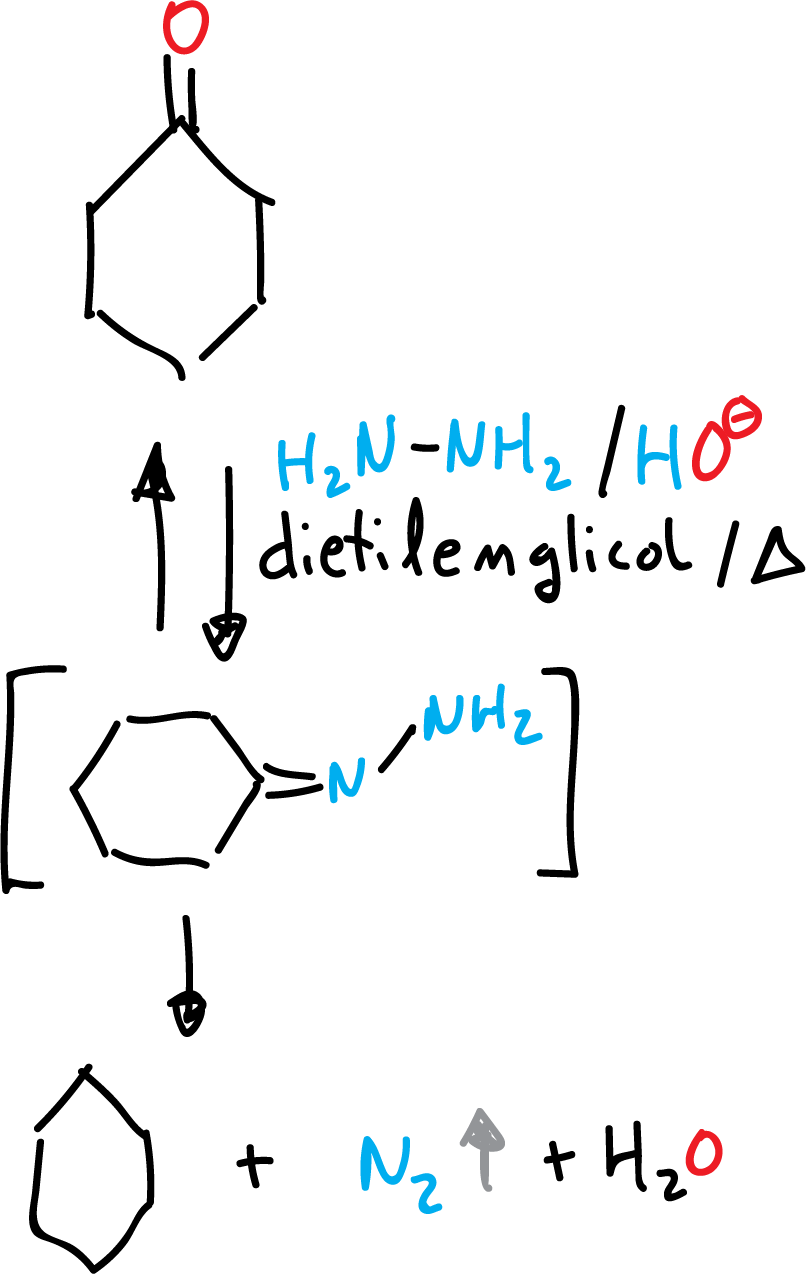
It is carried out under basic conditions (KOH), with excess hydrazine (NH2-NH2) and in high boiling point solvents (e.g. diethylene glycol has a boiling point of 245 ºC, HOCH2-CH2-O-CH2-CH2OH).
Water is formed, which must be removed to avoid lowering the boiling point The initial step is the formation of the corresponding hydrazone, followed by hydrogen migration and decomposition of N2.
Reduction to alcohol with metal hydrides (NaBH4 or LiAlH4)
Most aldehydes and ketones react readily with hydride transfer reagents such as metal hydrides. The two commonly used reagents are LiAlH4 and NaBH4, as a source of four hydride (H–) ions.
Due to their different structure, aldehydes generate primary alcohols and ketones secondary alcohols. The process takes place in two steps:
fig5
First, the hydride (nucleophile) is added to the carbonyl carbon (electrophile), the negative charge is displaced from the carbon to the oxygen forming an alkoxide.
The corresponding alcohol is then formed by protonation (H3O+). In ketones with different substituents a chiral center originates in the alcohol and the product is the racemic mixture of the R and S enantiomers.
Reduction to alcohol by catalytic hydrogenation
Hydrogenation, with transition metal catalysts (Ni, Pd or finely divided Pt), is the method commonly employed to reduce aldehydes and ketones on an industrial scale.
Aldehydes give rise to primary alcohols and ketones to secondary alcohols. If the ketone has different substituents, a chiral center arises and the product will be a mixture of the R and S enantiomers.
fig6
Addition
Addition of water (formation of geminal diol)
Aldehydes and ketones react with water to give 1,1-geminal diols called hydrates. In general, these hydrates are not stable, since the reaction is reversible and the starting reagents are formed. This reaction proceeds by acid or basic catalysis.
fig7
Addition of alcohol (formation of acetal)
Aldehydes and ketones react with 2 moles of alcohol to give 1,1-geminal diethers, commonly known as acetals. The term acetal is used interchangeably to describe aldehyde derivatives (acetals) and ketone derivatives (ketals).
It is usually performed under the following conditions: excess alcohol (ROH) solvent (benzene) and p-toluensulfonic acid (p-Ts-OH) as catalyst. They are useful as a protective group and in addition, 1,2- or 1,3-diols can be used to obtain cyclic acetals. These can be converted back to aldehydes and ketones by hydrolysis in acid medium or by iodine in methanol.
fig8
fig9
Thiol addition (thioketal formation)
Thioketals are the analogues of acetals, but with sulfur. They are obtained in the same way as acetals, but by reacting the carbonyl compound with thiol instead of alcohol. The carbonyl compound is regenerated by treatment with mercuric salts.
fig10
Addition of hydrogen cyanide (formation of cyanohydrin)
Hydrocyanic acid (HCN) is a weak (but very toxic) acid, which hydrolyzes easily giving the cyanide anion (strong conjugate base and good nucleophile).
fig11
This acid is generated in situ from NaCN or KCN with HCl. The cyanide anion attacks the carbonyl giving the cyanohydrins.
fig12
Ketones are more sterically hindered than aldehydes and the equilibrium shifts towards the reactants (low yield). It is a reaction that may be of synthetic interest because the cyano group can subsequently be converted to other useful functional groups such as -CO2H or -CH2NH2.
fig13
Ylide addition (Wittig reaction)
It is a reaction widely used in synthesis to form alkenes from a carbonyl compound with an ylide (phosphorus-stabilized carbanion). The ylides are neutral molecules, but with a charge separation.
They are obtained (in 2 steps) by reaction of a trialkyl (or aryl) phosphine with a haloalkane and subsequent acid-base reaction of the phosphonium salt with a strong base such as an alkyl lithium which generates the ylide.
fig14
The ylide can be described by the contribution of two resonant structures (one with a P=C double bond and the other with charge separation), the latter being favored (so that the carbon is nucleophilic against the carbonyl).
fig15
In the Wittig reaction, upon reaction of the phosphorus ylide with the carbonyl group, a structure called betaine is formed, which subsequently cyclizes to a four-membered ring (oxaphosphetane).
This ring is broken to generate a C=C (alkene) and a P=O bond, the stability of the latter being the driving force of the reaction. It also allows the double bond to be introduced without the problems of elimination reactions.
fig16
Addition of organometallic compounds
These are very useful C-C bond formation reactions in organic synthesis. Grignard (R-MgX) and organolytic (RLi) reagents are mainly used.
These reagents act as nucleophiles and are added to the carbon of the carbonyl group forming an alkoxide, which by protonation gives the corresponding alcohol. Ethers (Et2O or THF) are used as aprotic solvents.
The substituents of the aldehyde or ketone determine the type of alcohol obtained (formaldehyde gives primary alcohols, aldehydes give secondary alcohols, and ketones give tertiary alcohols).
The reaction ends with a stage where the acidic medium (H3O+) protonates the metal alkoxide salt.
fig17
Addition of amine and related compounds
Ammonia (NH3) and primary amines (R”-NH2) react with the carbonyl group to give carbinolamines, which are subsequently dehydrated to give imines (Schiff bases).
These reactions are still nucleophilic additions to the carbonyl, although they usually fall into a large group called condensations (two or more molecules join together, giving off water or another smaller molecule).
fig18
Secondary amines (R”R-NH) also react with aldehydes and ketones to give carbinolamines. Unlike primary amines, since the nitrogen proton is not available, in the subsequent dehydration step an enamine (C=C) is formed instead of an imine(C=N). Enamines are useful reagents in synthesis.
They also condense aldehydes and ketones with amine derivatives of general formula Z-NH2 such as hydroxylamines, hydrazines, phenylhydrazine, semicarbazide, giving oximes, hydrazones, phenylhydrazones and semicarbazones, respectively. These reactions are used for the identification of aldehydes and ketones.
Reactions at the α-carbon
Halogenation at the α-carbon
Halogenation of carbonyl compounds occurs via molecular halogen in a basic medium and yields the substitution of one or more α-hydrogens.
Normally, multiple halogenation products are obtained because chlorinated compounds are more reactive (the halogen removes charge and stabilizes the enolate anion).
In halogenations, the base does not act as a catalyst, so here it is used stoichiometrically. Depending on the substituents of the aldehyde or ketone racemates can be formed.
fig19
Haloform reaction
When a methylketone reacts with molecular halogen in the presence of a base, haloform (HCX3) is formed as one of the reaction products.
fig20
In acidic media the reaction takes place in a controlled manner, giving only the product of monohalogenation.
fig21
In contrast to ketones, aldehydes do not give good yields in α-halogenation because they are easily oxidized to carboxylic acids.
fig22
Other reactions
Reductive amination
It involves the transformation of an aldehyde or ketone into an amine. It is performed by treatment of the carbonyl group with a primary amine that generates an imine, which is reduced to an amine.
fig23
Cannizzaro reaction
It consists of the simultaneous autooxidation and reduction of the aldehyde, induced in a basic medium, of an aldehyde without hydrogens in the α-position, producing an alcohol and a carboxylic acid (in the form of the corresponding salt).
fig24
Back to the Synthesis and Reactivity of Organic Compounds.
Full Professor of Organic Chemistry at the University of Granada, with a long-standing research career in Computational Chemistry and molecular modeling and design.