
Go to the page with the list of problems.
Organic Synthesis – solutions to problems
The solutions to these Organic Synthesis problems are neither unique nor exclusive, the ones listed are some of the possible ones.
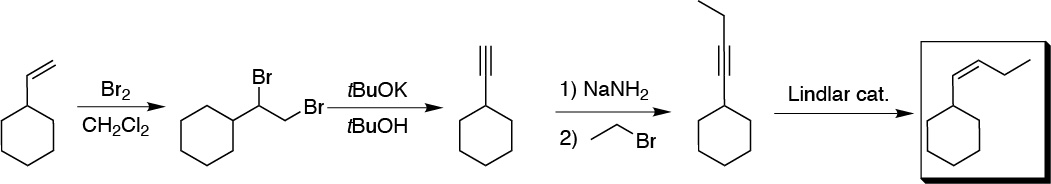
Solution 1:
The key to this synthesis is in the chain elongation, for this the chosen strategy is the conversion of the alkene to alkyne via the dibromo derivative, transformation to acetylide, nucleophilic substitution of a haloalkane and hydrogenation with Lindlar catalyst to give the alkene with the appropriate stereochemistry:
Solution 2:
The fastest and most convenient way to convert 2-methylchlorocyclopentane to 1-methylchlorocyclopentane is via the alkene:
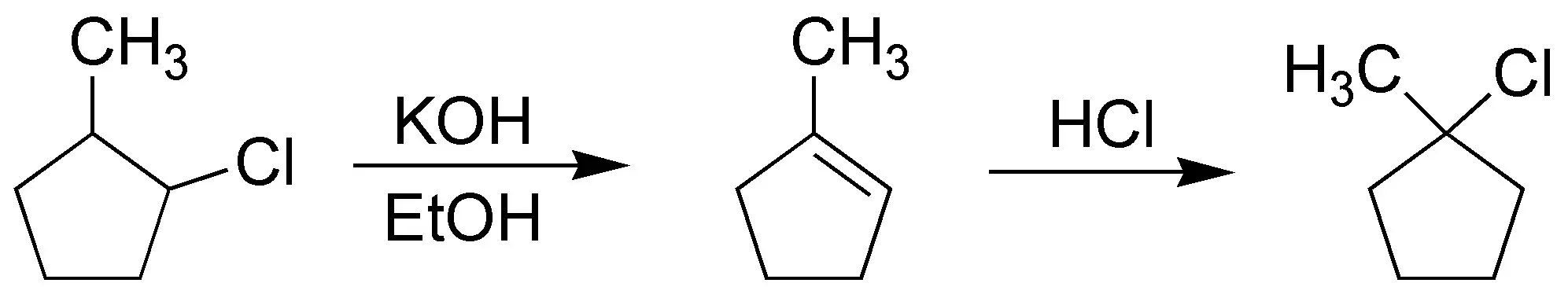
Solution 3:
In order to get the ketone group to convert to alkene (Wittig reaction) we must protect the aldehyde group which we do by acetalation reaction. Once protected, if we can use the Wittig reaction and subsequently deprotect the acetal:
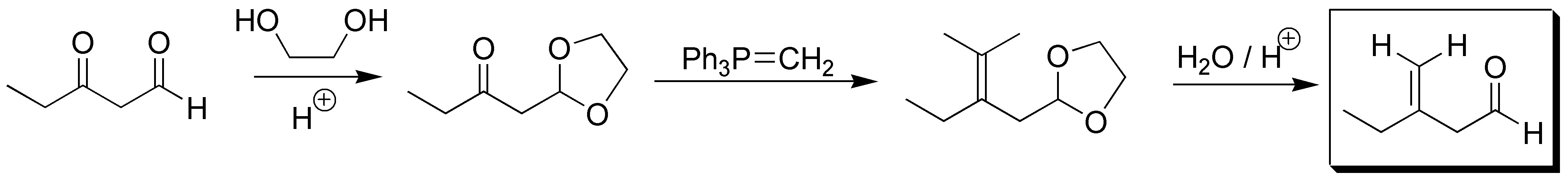
Solution 4:
First we proceed to lengthen the chain by means of a Grignard reaction, we convert the alcohol obtained into a new magnesian, after conversion to haloalkane. Carboxylation of this produces the corresponding carboxylic acid:
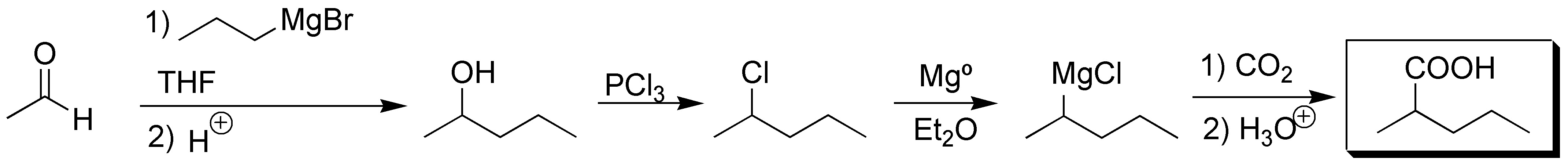
Solution 5:
Ozonolysis of the starting product gives the corresponding dialdehyde which by Wittig reaction with the corresponding phosphorus ylide we obtain the desired product:
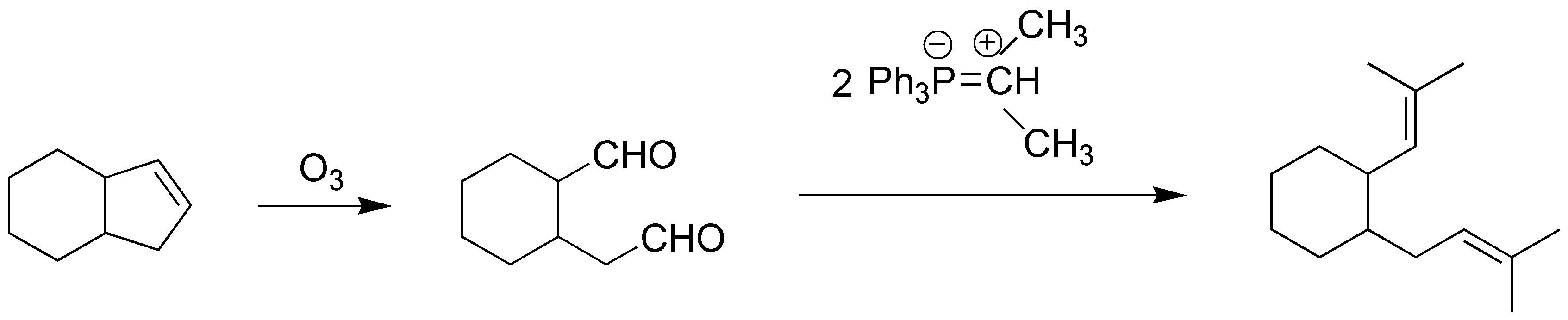
Solution 6:
1-propanol is converted to 2-bromopropane via propene. This is converted to a phosphorus ylide by treatment with triphenylphosphine and base. The reaction with propanone gives the expected alkene:
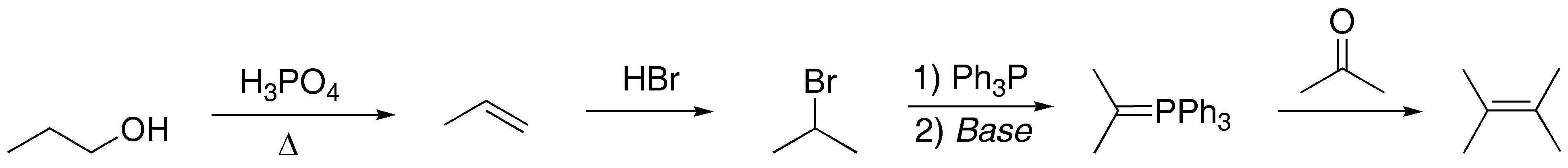
Solution 7:
Oxidation of the alkyl chain of an aromatic derivative leads to the corresponding carboxylic acid regardless of chain length. Reduction of the acid with LiAlH4 yields the corresponding alcohol.
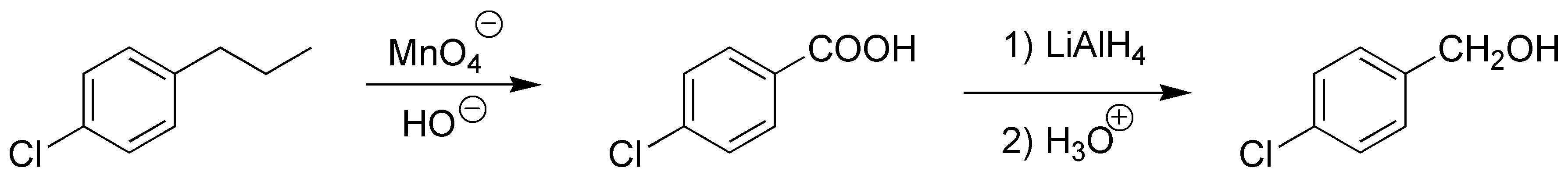
Solution 8:
Conversion of an alkene to aldehyde without degradation can easily occur upon conversion of the alkene to alkyne and subsequent anti-Markovnikov hydration of the alkyne.
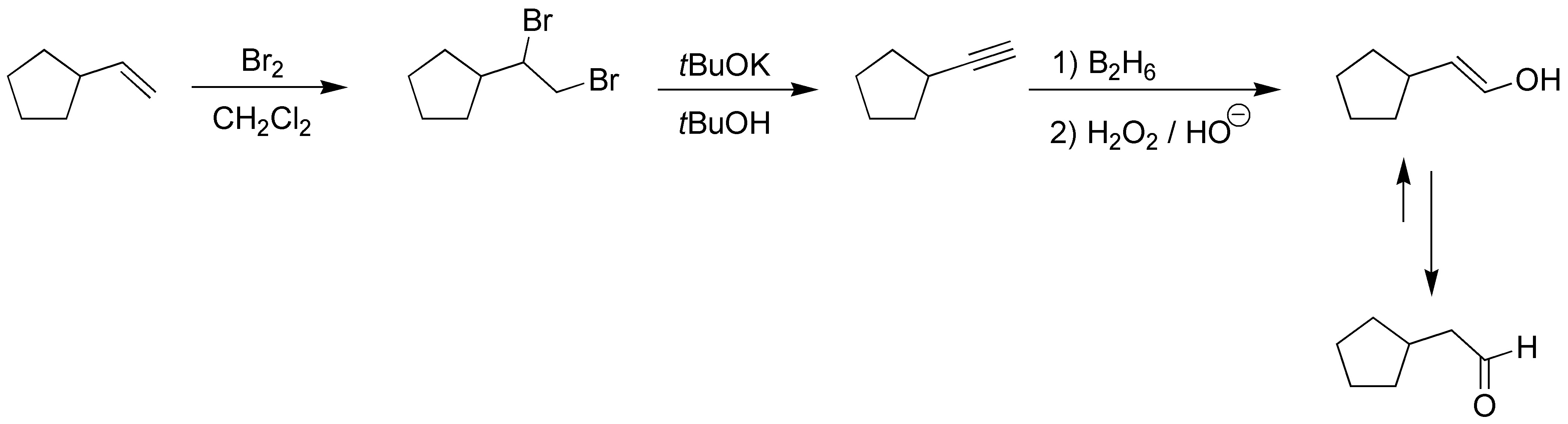
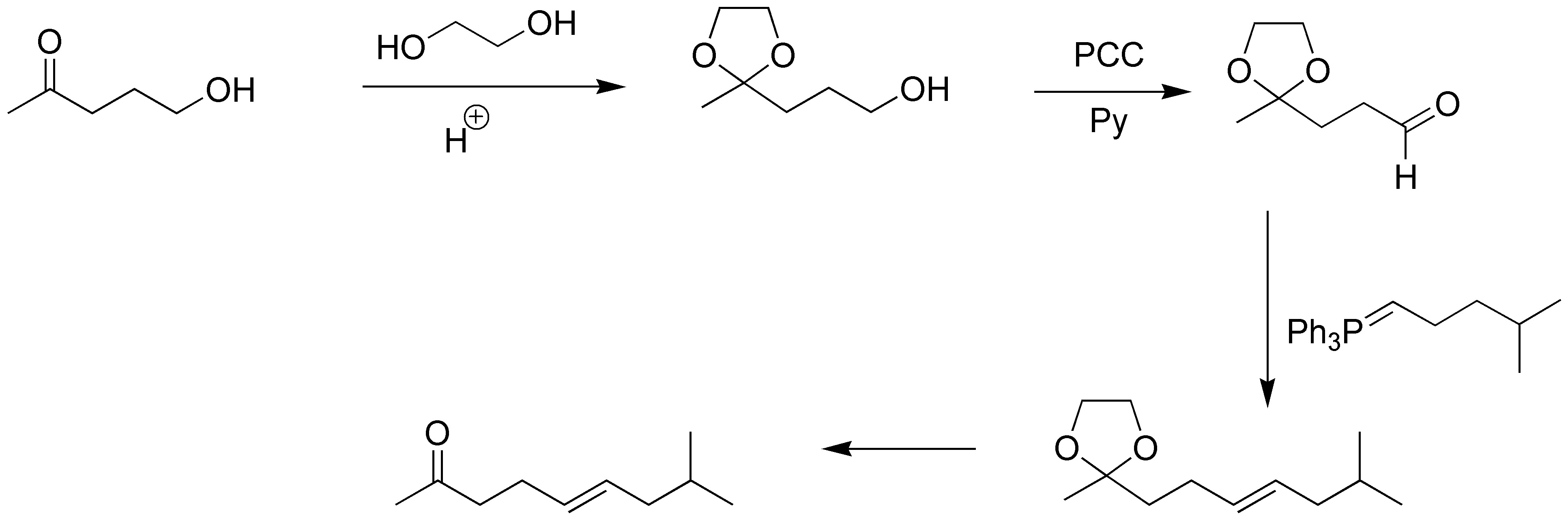
Solution 9:
To proceed with the elongation of the hydroxyketone chain by the hydroxyl group we must first protect the carbonyl group by means of a cyclic ketal. Once this is formed, we proceed to the oxidation of the hydroxyl to aldehyde with PCC in the absence of water, and subsequent Wittig reaction with the appropriate ylide gives an alkene. Finally we deprotect the ketal group by acid hydrolysis giving the desired ketone.
Solution 10:
The construction of a cyclohexene from an alkene suggests the use of the Diels-Alder reaction with a suitable diene: 2-methyl-1,3-butadiene. After separation of the two possible adducts formed, the major one is treated with an ylide to give the desired product.
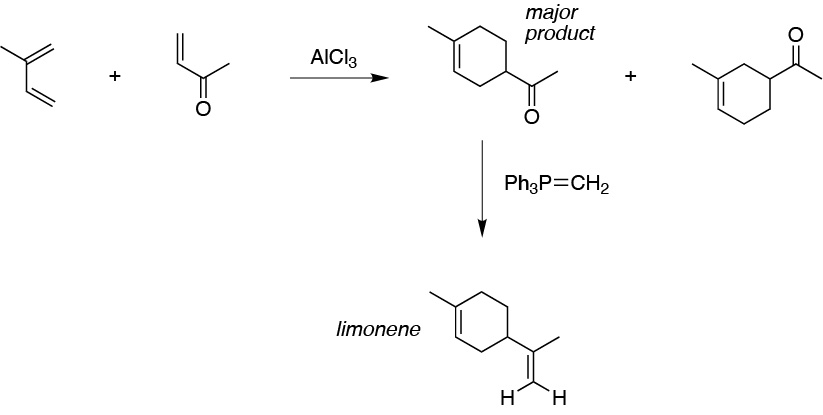
Solution 11:
Assuming that the double bond of the final product comes from the original triple bond, we must lengthen the chain on both sides of the acetylene by treating the corresponding acetylide with a suitable haloalkane. Hydrogenation with Lindlar catalyst or with Na / NH3 will give us the two desired isomers.
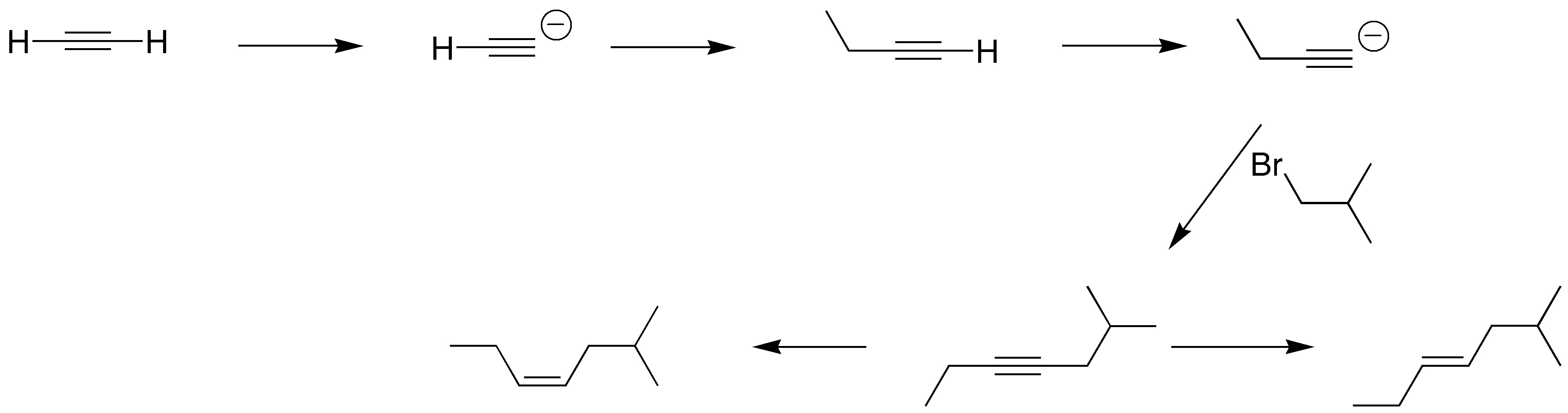
Solution 12:
Conversion of benzyl bromide to the corresponding Grignard compound and addition of this to acetaldehyde (ethanal) would give the corresponding secondary alcohol. Finally, by its oxidation the expected ketone is obtained.

Solution 13:
Halobenzenes also give Grignard compounds, when these are added to epoxides (oxacyclopropanes) they form alcohols (with ethylene oxide or oxacyclopropane) primary alcohol. Oxidation of the same to acid, conversion to acid chloride and treatment with ammonia generates the corresponding amide.
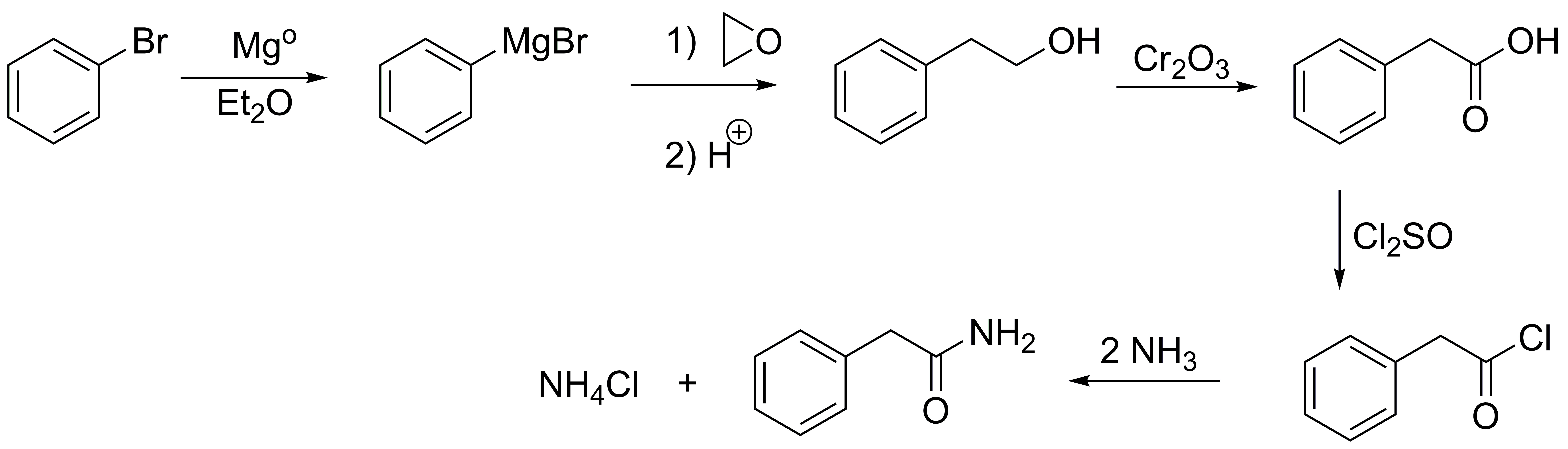
Solution 14:
A) The molecule in the exercise has two functional groups (double bond and carbonyl group). For transformation A, it is necessary to reduce the double bond of the ring leaving the carbonyl group unchanged (chemoselective reaction). A good option is to treat the substrate with hydrogen and a catalyst such as Pd on carbon. To avoid reduction of the carbonyl group, mild reaction conditions should be used, such as H2 pressure not exceeding 1 atm and room temperature.
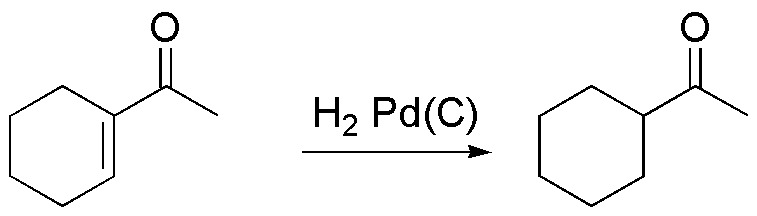
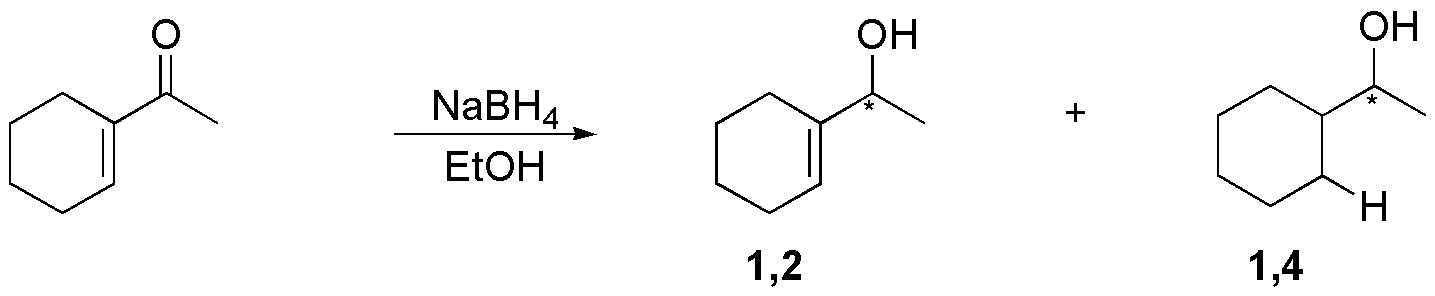
B) The reaction of the substrate with LiAlH4 and subsequent acidification of the reaction crude allows to reduce only the carbonyl group in the presence of the double bond (chemoselective reaction). When NaBH4 is used in conjugated systems, the carbonyl reduction product is usually accompanied by a certain amount of addition product 1,4 (reduction of the two functional groups).
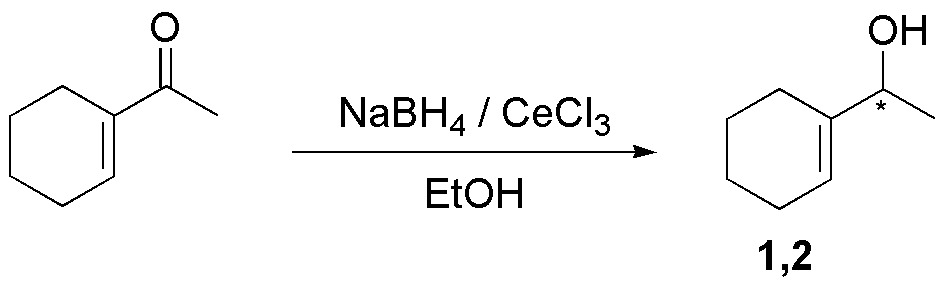
To make the reaction completely chemoselective with respect to the carbonyl, a variant is used for enones consisting of the addition of CeCl3 (Luche reaction).
C) The simultaneous reduction of the double bond and the carbonyl group can be carried out by hydrogenation, using an energetic catalyst such as Pt and H2 pressure between 3 and 5 atm.
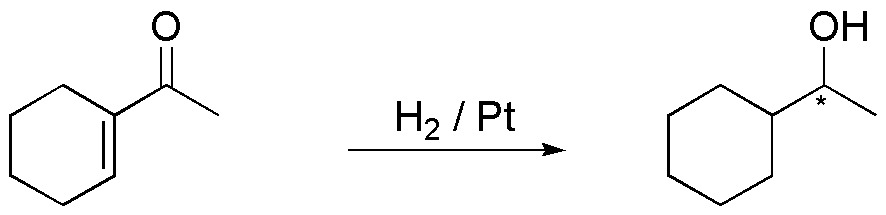
D) The reaction described is a conjugate addition of a methyl group. A nucleophilic carbon is required to carry out this reaction, such as organometallics. However, not all organometallics are useful for the reaction, as Grignard reagents and Li derivatives mostly yield the 1,2 addition product, while organocoumarates mostly give the 1,4 addition product.
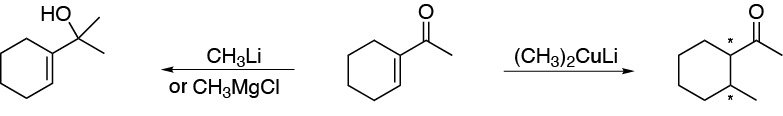
Solution 15:
Pentyl bromide with magnesium forms pentylmagnesium bromide, A, which reacts with ethylene oxide. Because the epoxide is symmetrical, a single opening product is obtained. The initial product of the reaction is an alkoxide, which by treatment with the aqueous acid will give the corresponding alcohol, B, with a carbon skeleton of two more carbons than the starting substrate. Oxidation with PCC of B (primary alcohol), leads to the formation of heptanal, C. The heptanal adds phenyl magnesium bromide, to give a secondary alkoxide, which in aqueous acid medium gives the alcohol D.
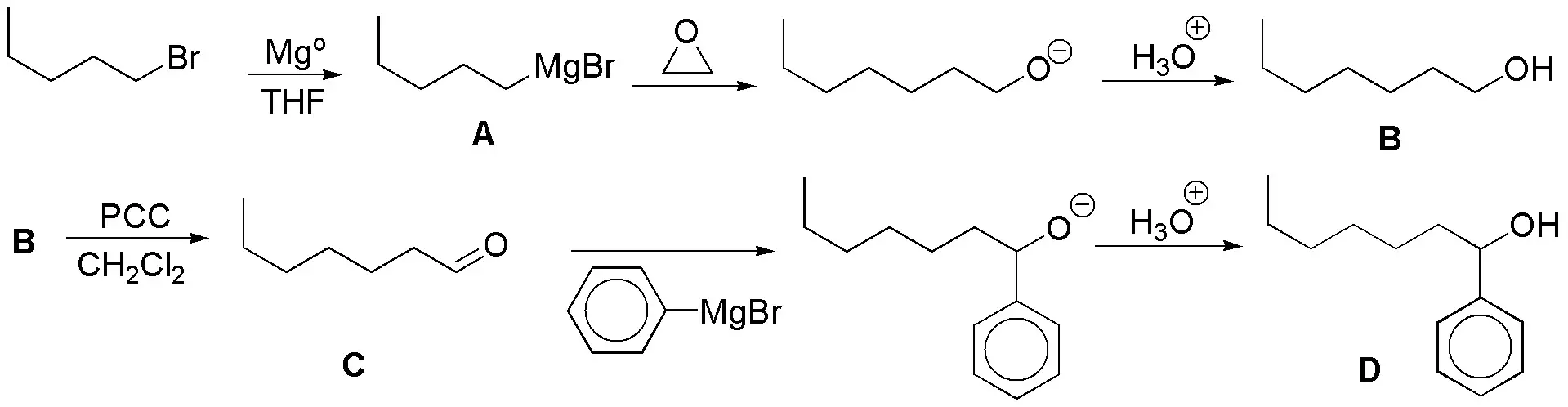
Solution 16
The comparison of the initial and final structures tells us that we must introduce two carbon atoms, due to the reactivity of the aldehyde group the first thing will be to protect this aldehyde as an acetal and then through a magnesian lengthen the chain with an aldehyde giving a 5-hydroxyacetal (E) that after deacetalization gives the hydroxyaldehyde (F) can be found in equilibrium with the hemiacetal form (K), that with a mild oxidation with bromine in water would give the corresponding lactone. More complex is the proposed synthesis in which we proceed with oxidation of F to the corresponding ketoacid, selective reduction of the ketone group and esterification giving J:
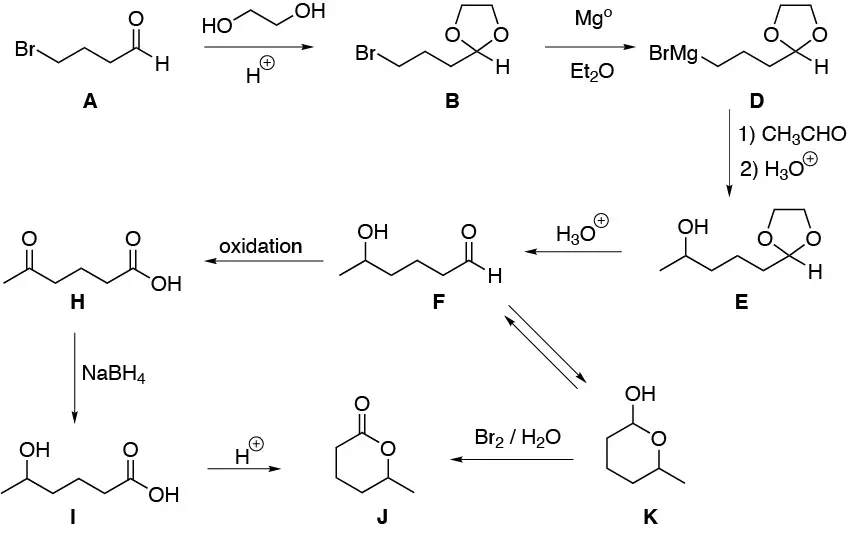
Solution 17:
We convert the ketone to the corresponding ketal and by allylic bromination of the resulting alkene we obtain the 4-bromo derivative. The formation of the magnesian and carboxylation of the same would give us the corresponding carboxylic acid salt. Finally, by acid hydrolysis of the same we would obtain the desired product.
Solution 18:
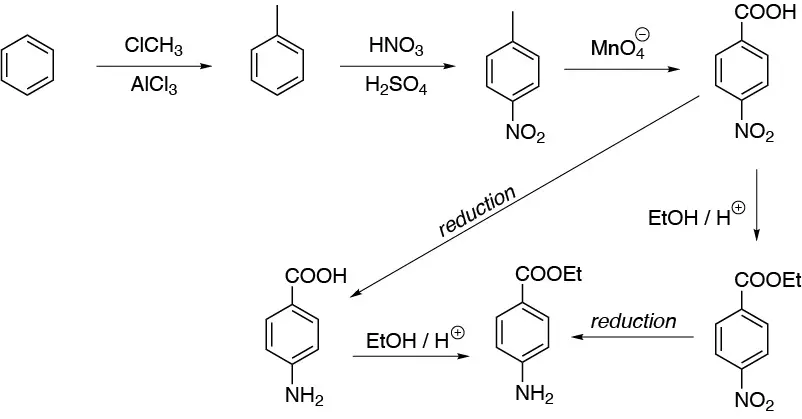
Alkylation and subsequent nitration of benzene yields 4-nitrotoluene, the oxidation of which gives us 4-nitrobenzoic acid. From this we can esterify with ethane and reduce the nitro group or reduce the nitro group and esterify the acid. Both methods produce the desired product.
Solution 19:
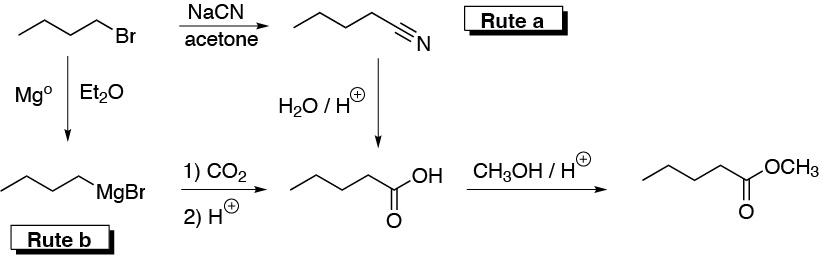
To obtain pentanoic acid we are presented with two possible routes: a) Cyanide treatment and subsequent hydrolysis of the nitrile formed or b) formation of the magnesian and carboxylation of the same. Subsequent esterification of the same would give us the desired ester.
Solution 20:
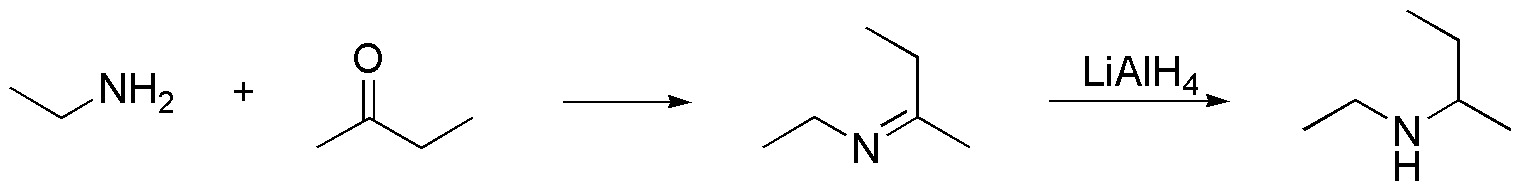
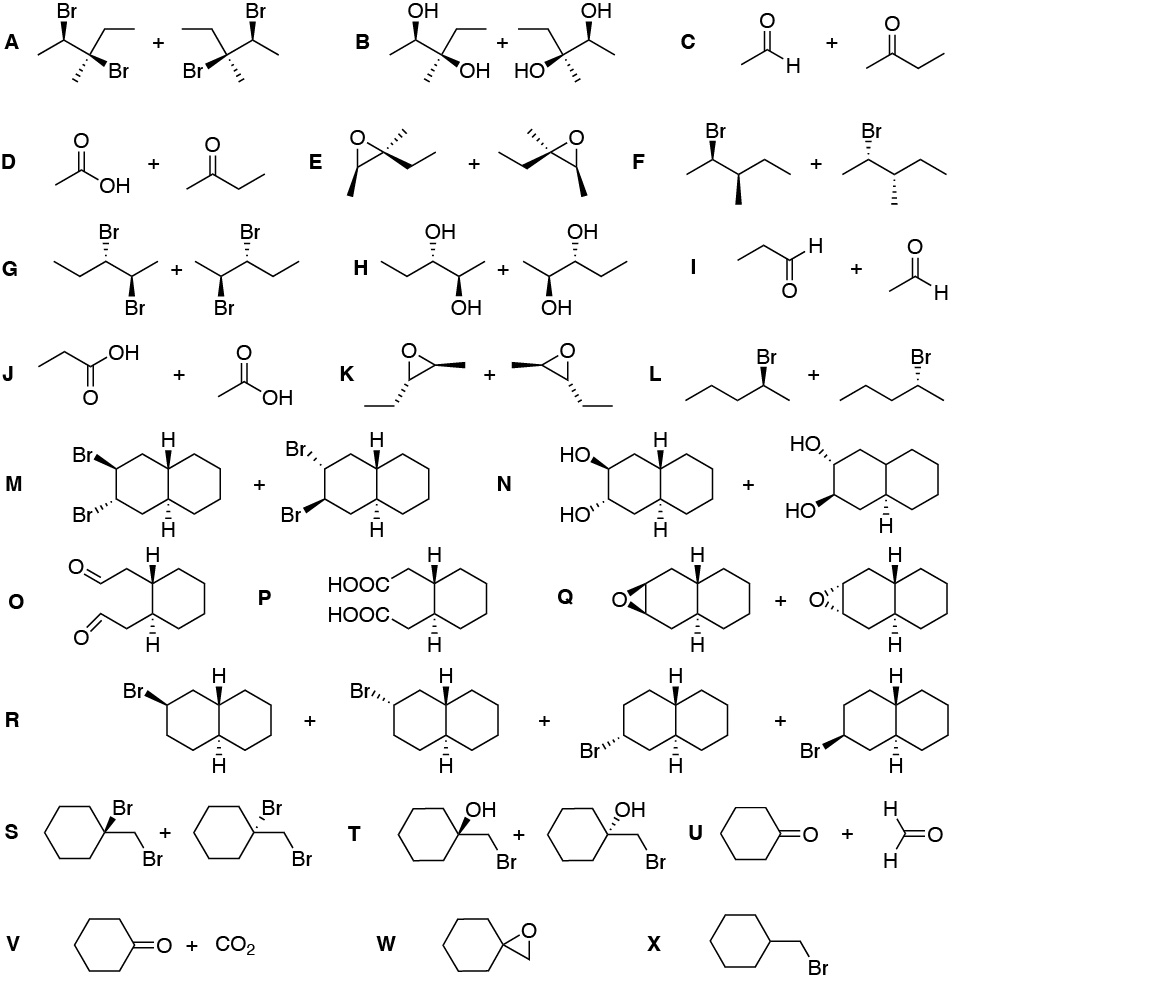
a) A first consideration is that there may be several solutions to the same problem, since the way of assemblingfragments to obtain a given molecule is not unique. In this case, we divide the molecule in two different zones joined through the nitrogen atom.
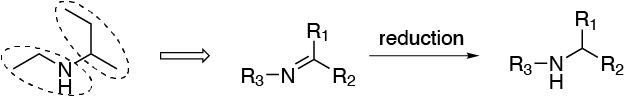
A good option may be to prepare an imine, such as the one in the figure, which, when reduced, leads to the formation of the secondary amine that is the target of the synthesis. As is known, imines are the result of the reaction between an amine and a carbonyl compound.
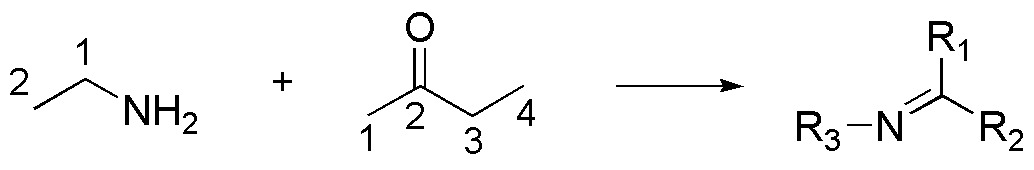
The amine has 2 carbon atoms and the ketone has 4. One possible route for the preparation of this fragment would be from the initial products or carbon sources. This could be achieved from methanol, transforming it into a halogenated derivative, increasing the carbon chain by displacement with cyanide. The reduction of the nitrite would lead to the desired amine.
As for the four-carbon ketone, it can be synthesized from ethanol in several steps:
![]()
![]()
- Convert ethanol to propanol via a magnesian and formaldehyde (in two steps).
- Oxidize the primary alcohol to give propanal.
- Increase the carbon chain by one unit with methylmagnesium bromide (prepared from methanol, according to the scheme).
- Oxidize the secondary alcohol to ketone.
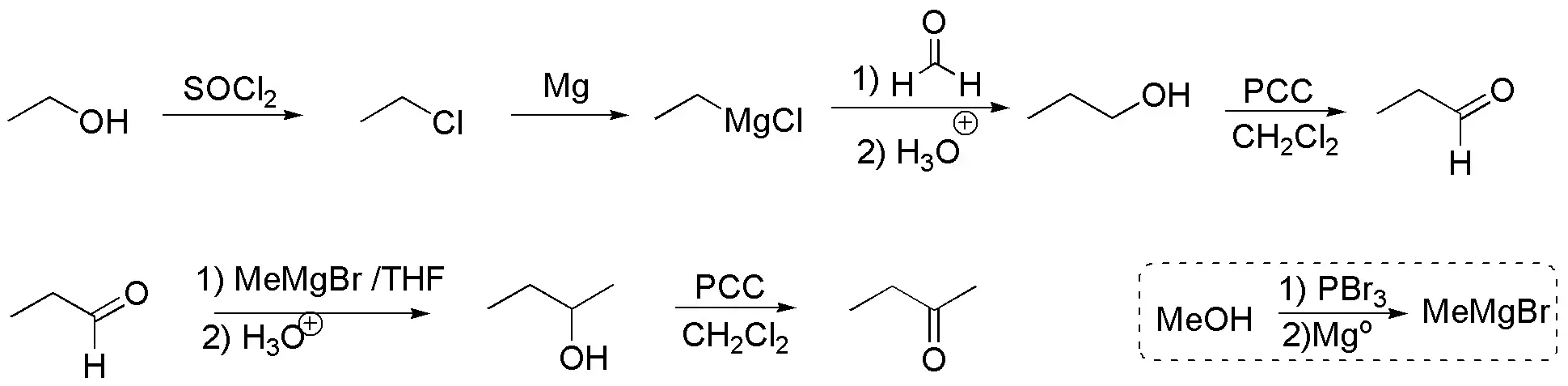
Finally, join both fragments, amine and ketone, to give the imine and subsequently reduce, e.g. with LiAlH4.
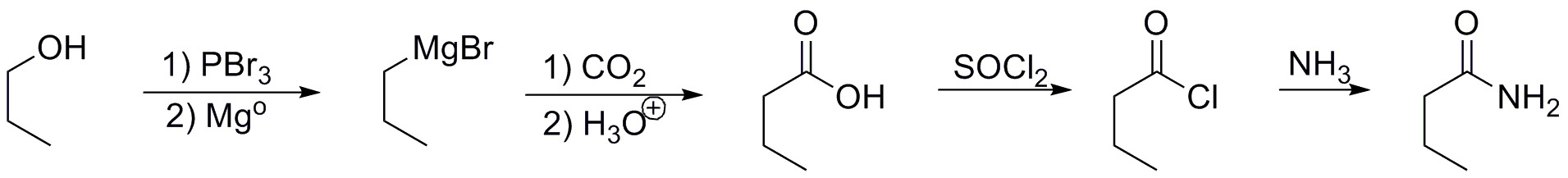
b) To prepare substrate b) a linear 4-carbon carbon skeleton is required. This can be started from propanol, which has been prepared from ethanol in the previous section. The propanol is converted in two steps to a magnesian which with CO2, which is another carbon source, forms butanoic acid.
The butanoic acid can be transformed into the corresponding acyl chloride by treatment with thionyl chloride. Finally, the acyl chloride reacts with ammonia to give the amide that is the subject of the synthesis.
Solution 21:
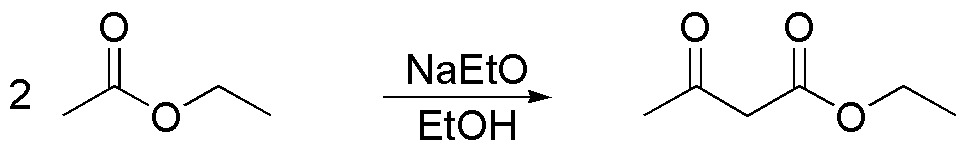
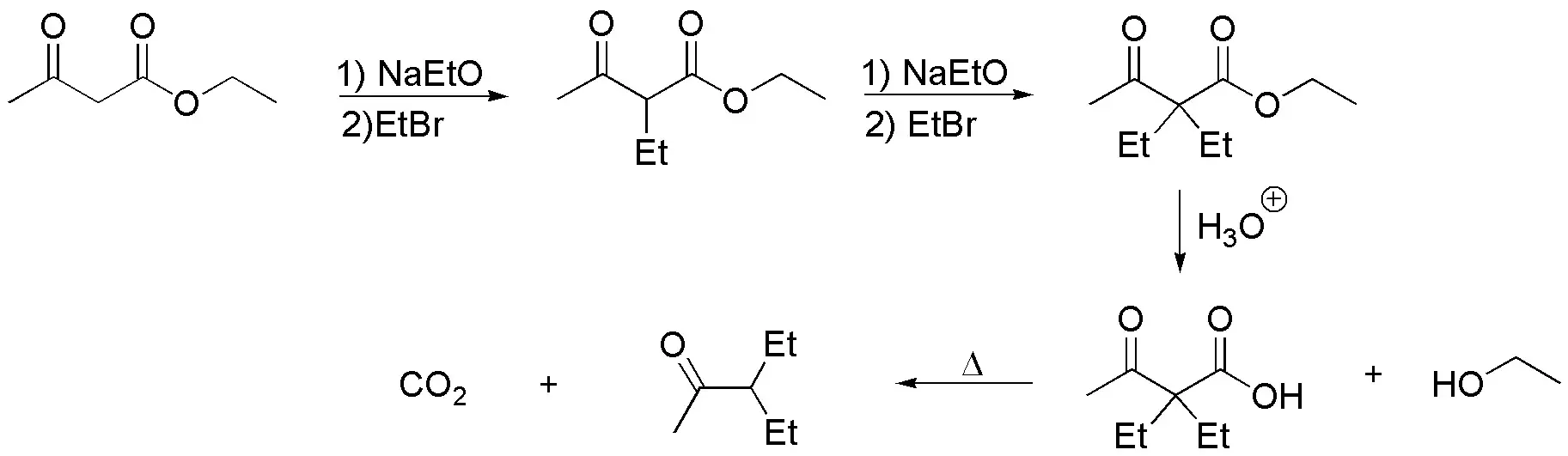
a) The molecule is a disubstituted methylketone in the position adjacent to the carbonyl group. The most usual procedure to prepare this type of compounds is by acetoacetic synthesis, which allows to obtain mono- or di-substituted methylketones in position 3 of the chain according to the sequence of reactions:
- Alkylation of ethyl acetoacetate.
- Hydrolysis of the ester.
- Decarboxylation.
so that the fragment appearing in the thick trace is contributed by the ethyl acetoacetate while the other fragments come from the alkylation reactions.
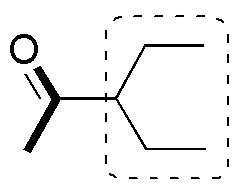
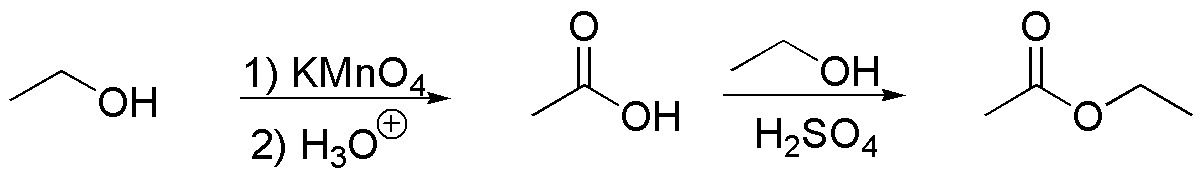
Since the only carbon source is ethanol, we must prepare ethyl acetoacetate in several steps from ethanol. Claisen condensation is a suitable procedure to obtain acetoacetate from ethyl acetate.
In turn, ethyl acetate is obtained from acetic acid and ethanol and acetic acid can be prepared by oxidation of ethanol.
The scheme of the acetylacetic synthesis would be as follows:
Finally, the ethyl bromide used, can be prepared from ethanol by any of the procedures described in the conversion of alcohols to haloalkanes.
![]()
![]()
b) A fragment (ethyl acetoacetate) can be distinguished in the molecule which can be assembled into the molecule through a C-C bond formation reaction to the rest of the molecule.
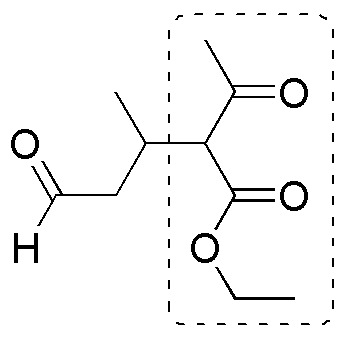
The key to this synthesis lies in the linkage between this fragment and the rest of the chain through a Michael addition of the carbanion of the ethyl acetoacetate onto the following unsaturated aldehyde:
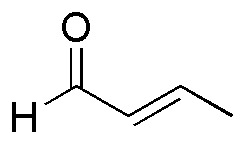
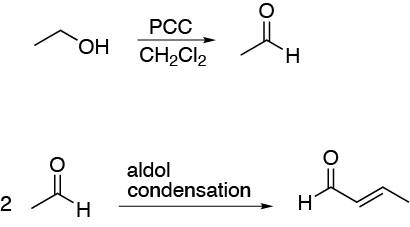
This Michael acceptor, can be prepared from ethanol according to the following sequence of reactions:
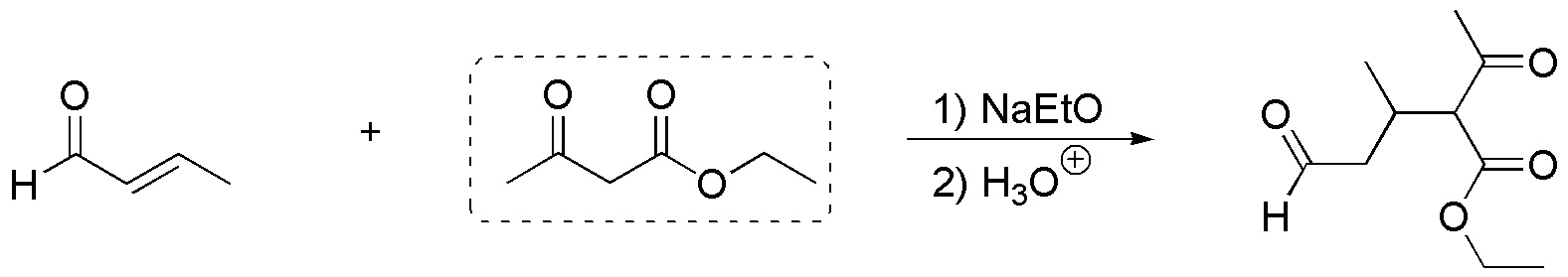
The reaction allowing the synthesis of compound b) is:
Solution 22:
The first stage of the malonic synthesis is, from the point of view of the mechanism of the reactions, an SN2-type process, in which a resonance-stabilized carbanion acts as a nucleophile and displaces a halogen atom in an alkyl halide. Bromides are almost always used, since Br is a good leaving group and its halides are relatively stable and easy to handle.
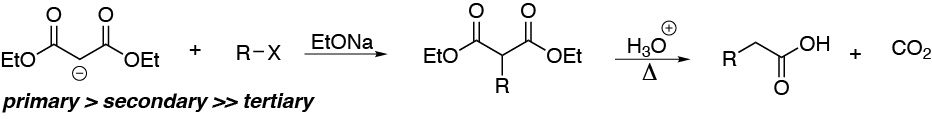
Therefore, depending on the nature of R, the reaction will proceed more or less easily. The halides needed are:

The ease with which the reaction occurs will be: I ≈ III > II
With IV the reaction is not feasible, since it is a tertiary halide. For compound V a different strategy must be followed, which is more effective. It consists of using a dihalide, with Br and Cl at the end of a carbon chain, with the same number of carbons as the cycle. Since Br is a better leaving group than Cl, the substitution reaction occurs first at this point. Subsequently, the intramolecular substitution reaction occurs to give the four-membered cycle. As in all cases, the reaction is completed by hydrolysis and subsequent decarboxylation.
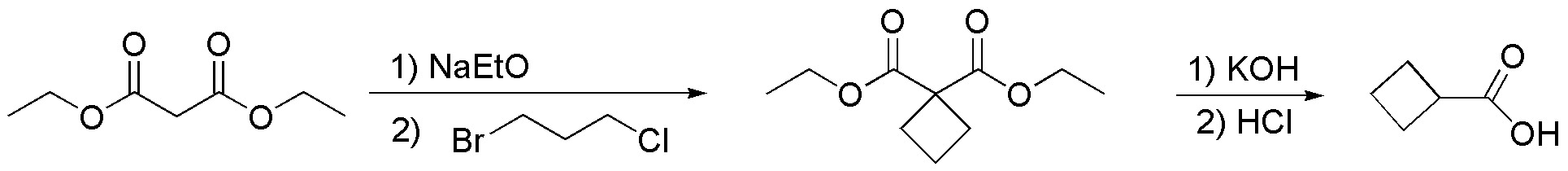
Solution 23:
First we calculate the number of unsaturations: as alkanes possess a molecular formula CnH2n+2, in this case hexane would be C6H14 the molecule possesses only one double bond. If in ozonolysis only propanal is produced, it means that the double bond is symmetrically substituted, and therefore it is hex-3-ene:
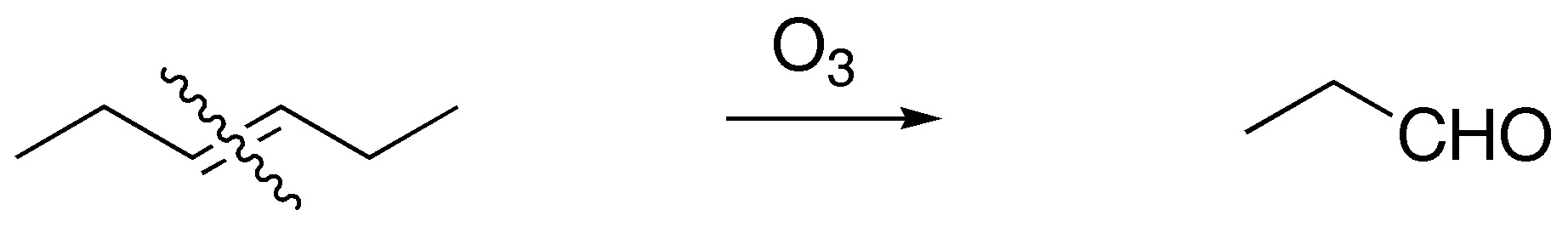
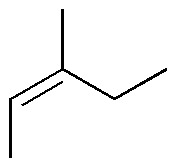
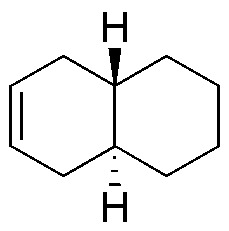
Solution 24:
| Br2 / Cl4C | H2O2 / OsO4 (cat.) | Ozonolysis | KMnO4 (hot) | Peracids | HBr / peroxides | |
|---|---|---|---|---|---|---|
 | A | B | C | D | E | F |
| G | H | I | J | K | L | |
 | M | N | O | P | Q | R |
| S | T | U | V | W | X |
The first column of the table corresponds to the addition of bromine to the double bond (anti addition) giving the corresponding derivatized dibromo. The second column is a dihydroxylation without the double bond giving the corresponding glycol. The third and fourth consist of oxidative cleavage of the double bond giving 2 carbonyl compounds and/or CO2. The fifth is the epoxidation of double bonds giving the corresponding oxacyclopropanes (epoxides) and the last is the anti-Markovnikov addition to the double bond:
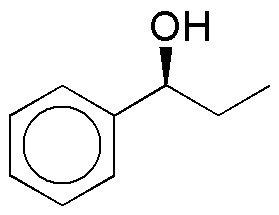
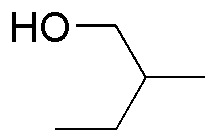
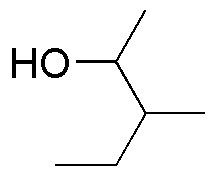
Solution 25:
| PCC /CH2Cl2 | CrO3 / H3O+ | KMnO4 | |
|---|---|---|---|
 | A | B | C |
 | D | E | F |
 | G | H | I |
PCC is a mild oxidant that oxidizes primary alcohols to aldehydes and secondary alcohols to ketones. Chromic oxide and potassium permanganate are strong oxidizers:
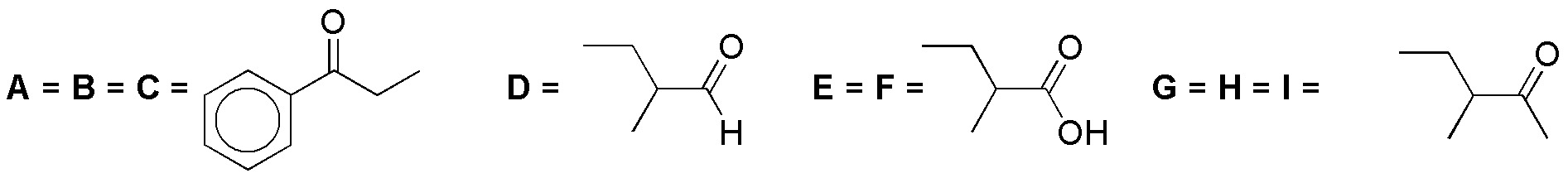
Solution 26:
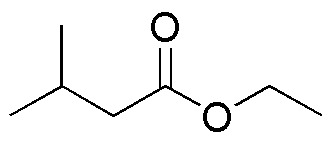
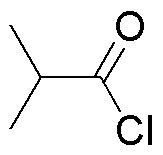
| LiAlH4 | NaBH4 | B2H6 | DIBAH | |
|---|---|---|---|---|
 | A | B | C | D |
| E | F | G | H | |
| I | J | K | L | |
 | M | N | O | P |
The table illustrates the behavior of different carbonyl compounds against different reductants.
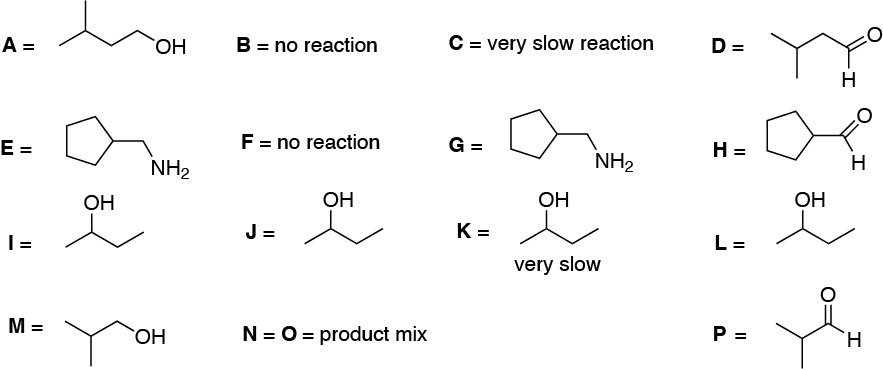
Solution 27:
The following scheme illustrates the main reactions of alkenes and the interconversions and relationships existing between the products formed in the direct reactions of alkenes.
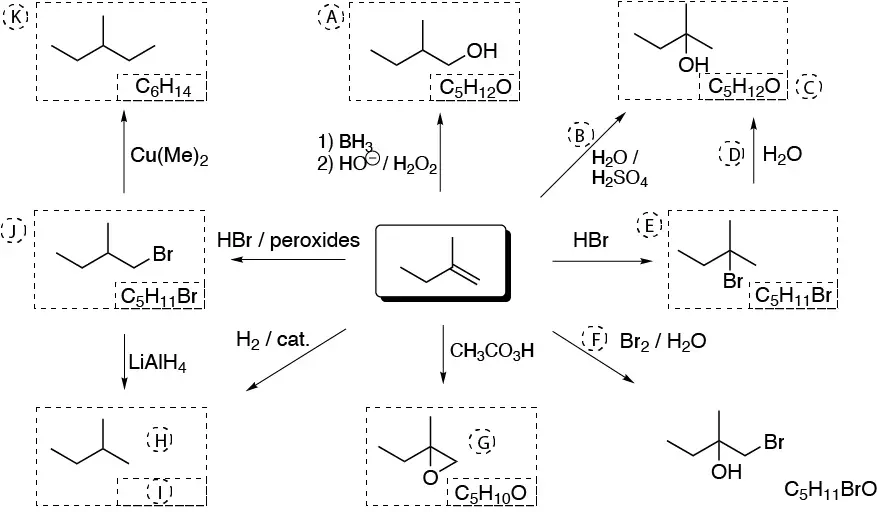
Solution 28:
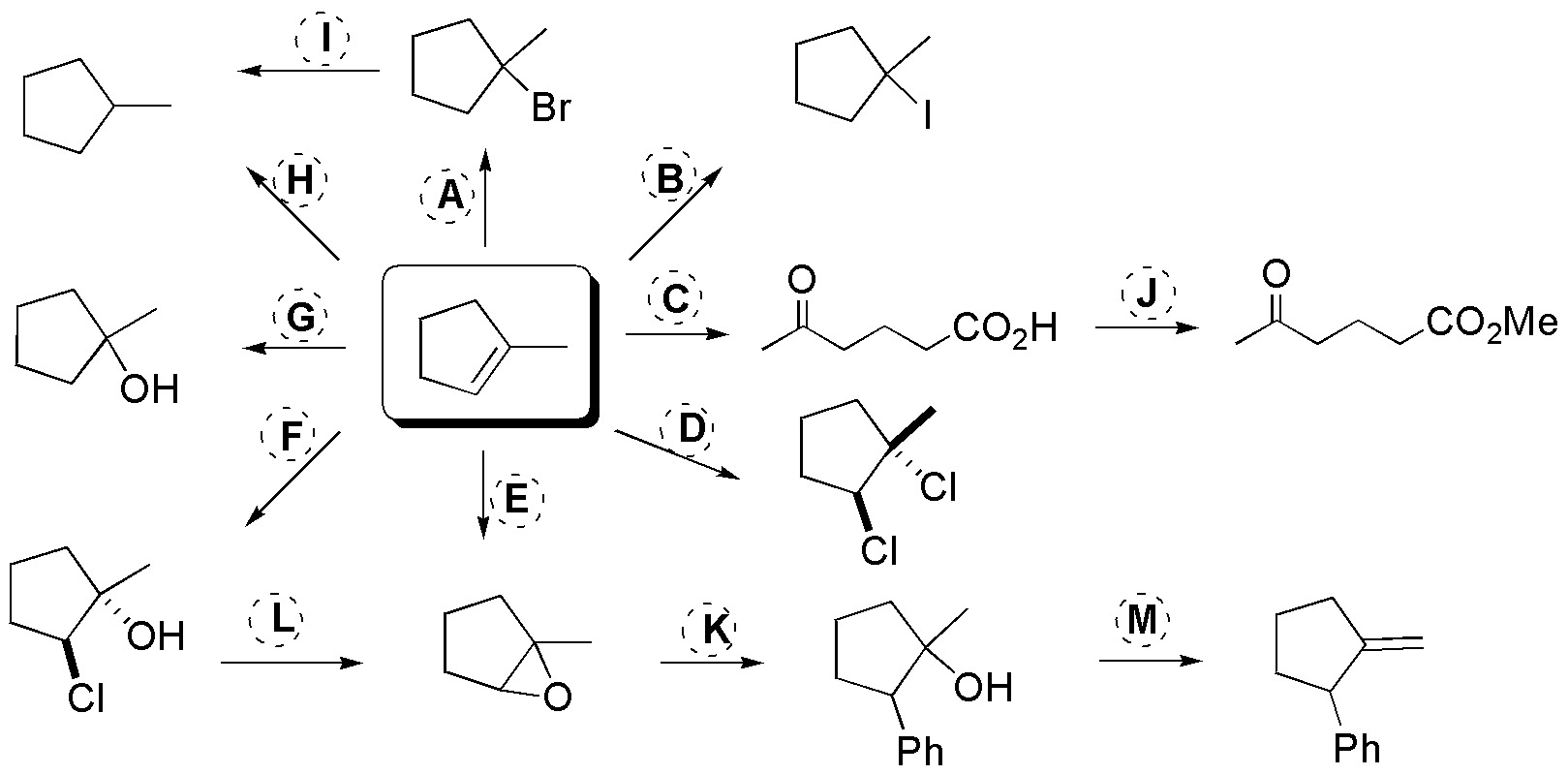
The following scheme illustrates the main reactions of alkenes and the applications of the products formed in the direct reactions of alkenes:
A = HBr; B = KI / H3PO4; C = 1) KMnO4 / HO–, 2) H3O+; D = Cl2 / CCl4; E = peracid; F = Cl2 / H2O; G = H2O / H2SO4; H = H2 / (cat.); I = (Bu)3SnH; J = diazomethane; K = PhLi / THF; L = NaH / THF; M = KtBuO / tBuOH.
Solution 29:
The first stage of the proposed synthesis is a hydration of alkenes with different anti-Markovnikov regioselectivities. The first and Markovnikov the second. The second stages correspond to the different oxidation conditions: with PCC to a carbonyl compound and with dichromate to acidic:
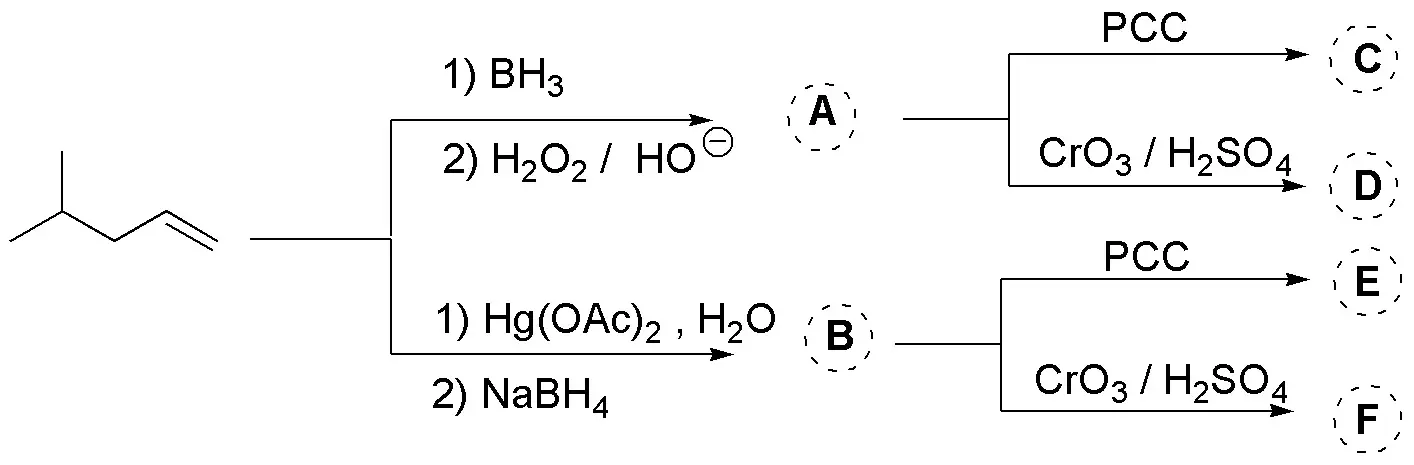
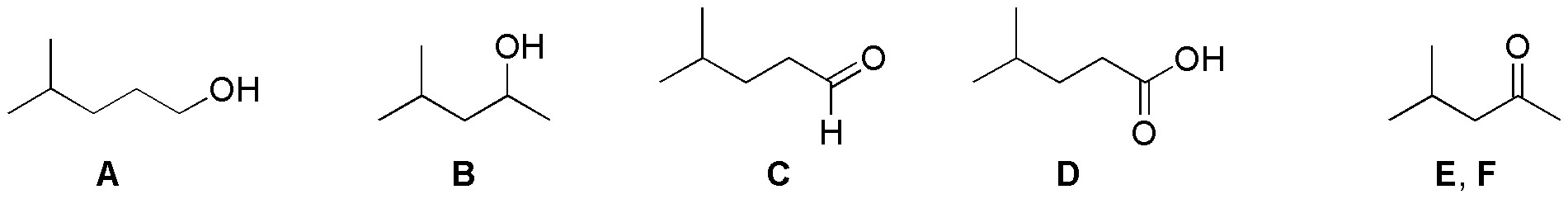
Solution 30:
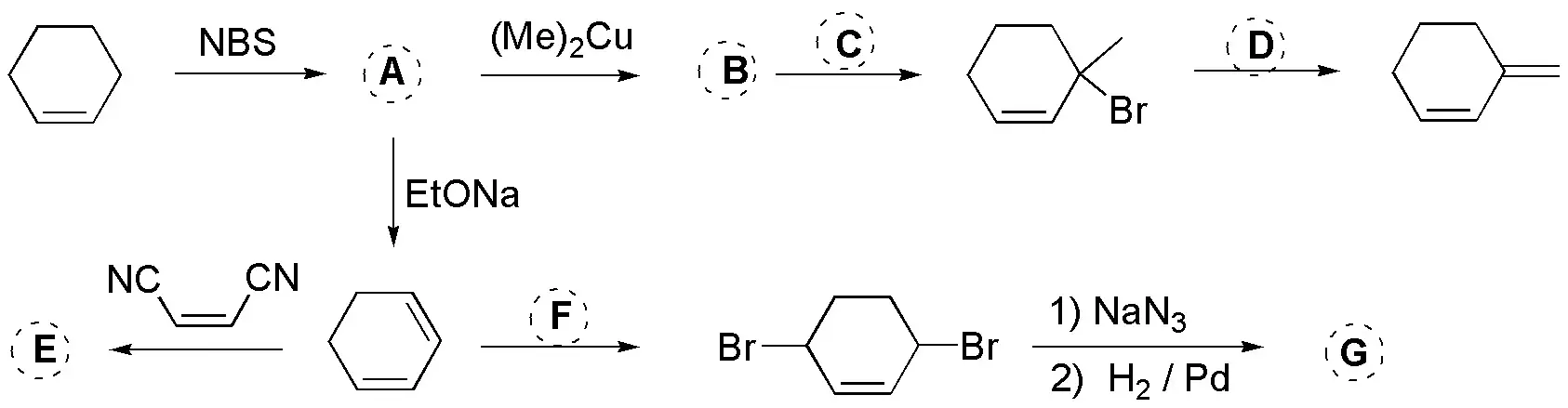
NBS produces the allylic halogenation of alkenes (A, 3-bromocyclohexene), treatment of this with a strong base will produce a diene and with an organocuprate the 3-methylcyclohexene, the radical halogenation of which will produce 3-bromo-3-methylcyclohexene. Treating this with a strong and hindered base (bulky) such as potassium tert-butoxide will produce the indicated diene. For its part, cyclohexadiene can give a Diels-Alder reaction with ethenodinitrile giving the bicyclic compound E or it can add bromine under thermodynamic conditions (1,4- addition) to give dibromocyclohexene. The latter, after undergoing a double nucleophilic substitution with azide and subsequent reduction thereof gives the diaminderivative G:

Solution 31:
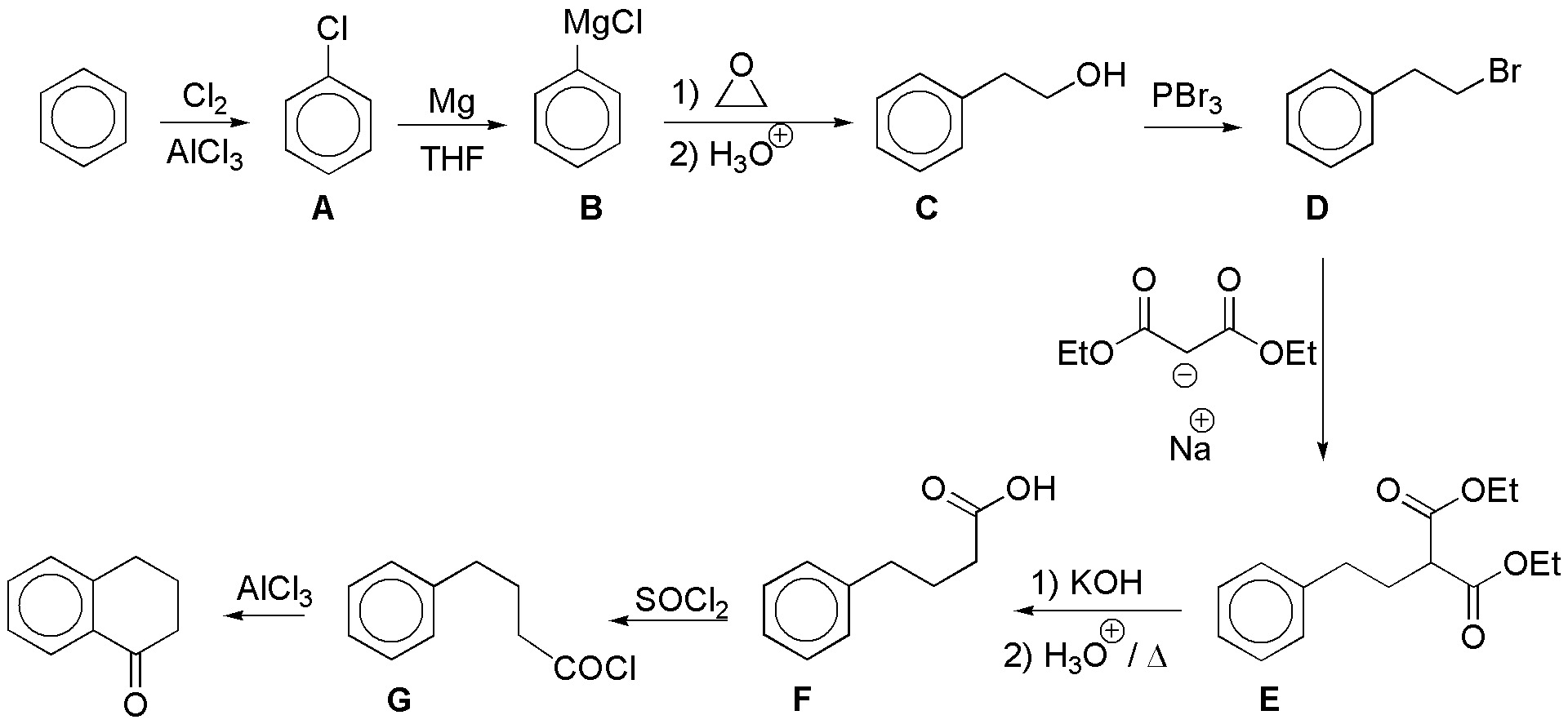
Chlorination of benzene yields chlorobenzene (A) which is transformed into a magnesian (B). This reacts with oxacyclopropane (ethylene oxide) producing the primary alcohol C, which after transformation into the bromoderivative D, allows alkylation of ethyl malonate giving E; this β-diester is saponified with decarboxylation by treatment with potash giving the corresponding carboxylic acid (F). To produce the cyclization by a Friedel-Craft acylation we need to convert it to the corresponding acid chloride G:
Solution 32:
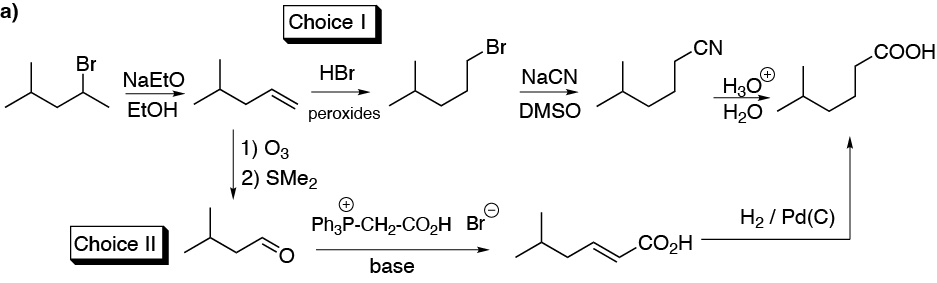
a) We need to lengthen the chain by one carbon atom, to do this the first thing is to transform the bromoalkane into an alkene, subsequently we will have two alternatives:
Route 1: Anti-Markovnikov addition of HBr to the alkene, substitution with NaCN and hydrolysis of the nitrile obtained giving the acid sought.
Route 2: Ozonolysis of the alkene, reaction of the aldehyde with the phosphorus ylide indicated in the scheme and hydrogenation of the double bond:
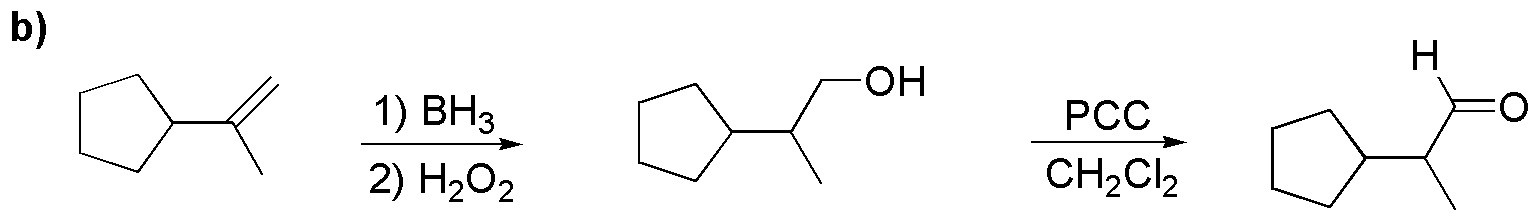
b) Anti-Markovnikov hydration of the alkene produces a primary alcohol, which with PCC can be oxidized to the corresponding aldehyde:
c) After bromination of the diphenylmethane. This is bromide reacted with the propynyl acetylide, giving the disubstituted alkyne. By hydrogenation with Lindlar catalyst we will obtain the corresponding alkene with the desired stereochemistry:
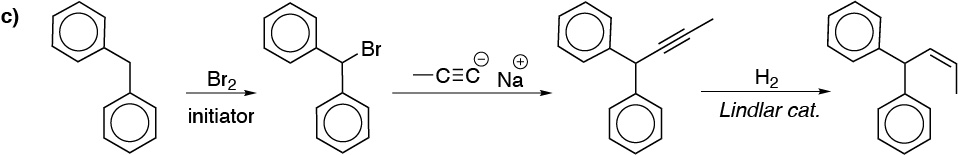
d) We need to introduce two non-consecutive carbon atoms into the initial structure. For this, the basic intermediate is propanone which is obtained by oxidation of 2-propanol. Alkylation of the enolate thereof leads to butanone, which by Wittig reaction transforms the desired alkene:
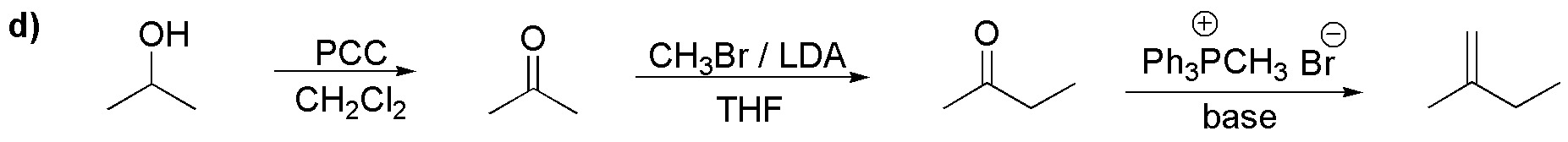
Solution 33:
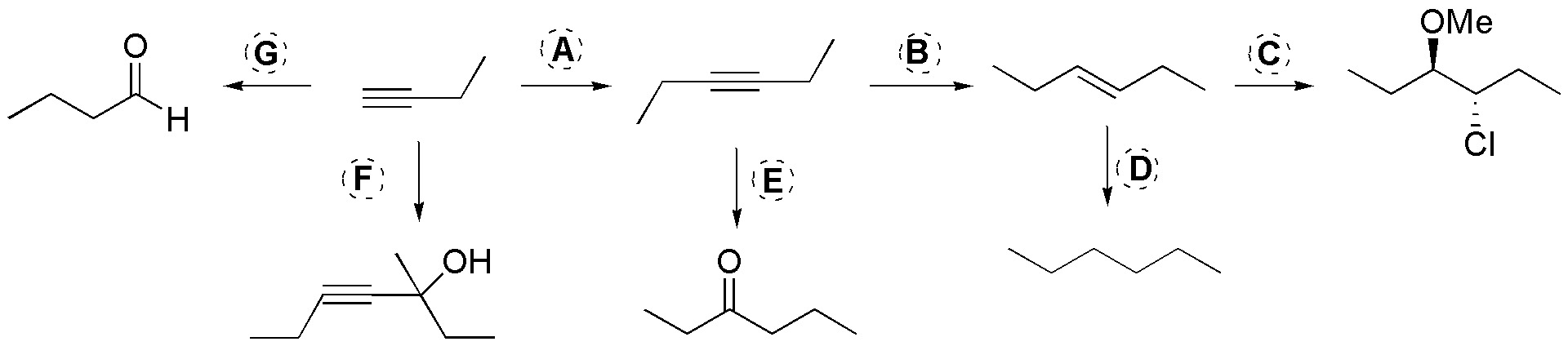
A) To lengthen the chain of a terminal alkyne we will use the sequence: 1) NaNH2 / THF, 2) EtBr; B) The hydrogenation of the alkyne to give trans stereoisomerism in the alkene occurs with: Li / NH3; C) It is about introducing Cl and OMe into the molecule for them we will do halogenation with chlorine in methanol; D) It will be a hydrogenation of alkene so we will use: H2 /Pd(C); E) The alkyne-ketone conversion is achieved by hydration of the alkyne (the intermediate will be an enol), as this is symmetrical no ketone mixtures will be formed, a possible solution will be: H2O / H3PO4; F) Acetylides can be nucleophilically added to ketones to produce alkynyl alcohols, therefore we will proceed: 1) NaNH2, 2) 2-butanone; G) The alkyne-aldehyde conversion is easily achieved by anti-Markovnikov hydration of the propyne, therefore the sequence used will be: 1) BH3, 2) H2O2 / HO–.
Solution 34:
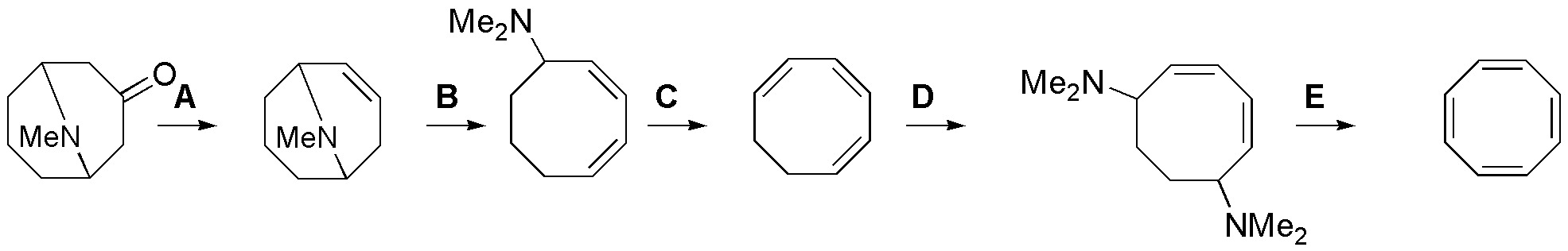
A) It will consist of a reduction of the ketone and subsequent dehydration of the alcohol, therefore: 1) NaBH4 , 2) H2O / H2SO4; B = C = E) These three steps consist of Hofmann eliminations so the sequence used is: MeI, Ag2O / heat; D) Since it is not possible to add the amines to the double bonds we must stop for the dibromo derivative and subsequent substitution of the same, therefore the sequence used will be: 1) Br2 / CCl4, 2) Me2NH.
Solution 35:
The formation of substituted cyclohexenes leads us to use the Diels-Alder reaction as a key step in the synthesis. For this we must convert the initial product into a substituted diene which we achieve through benzyl halogenation and subsequent elimination of the derived dibromo:
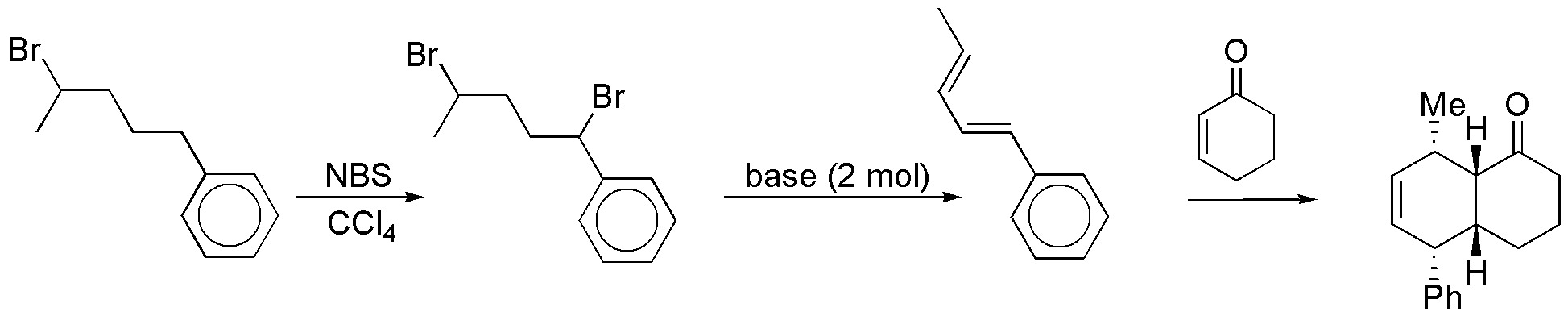
Solution 36:
A) It is a magnesian that could be added to epoxides nucleophilically on the least hindered carbon giving an alcohol (B). Transformation of the same to bromide and subsequently to magnesian and addition of the same to propanone will give us the unsaturated alcohol C. Anti-Markovnikov hydration of C will give us the desired product via borane D:
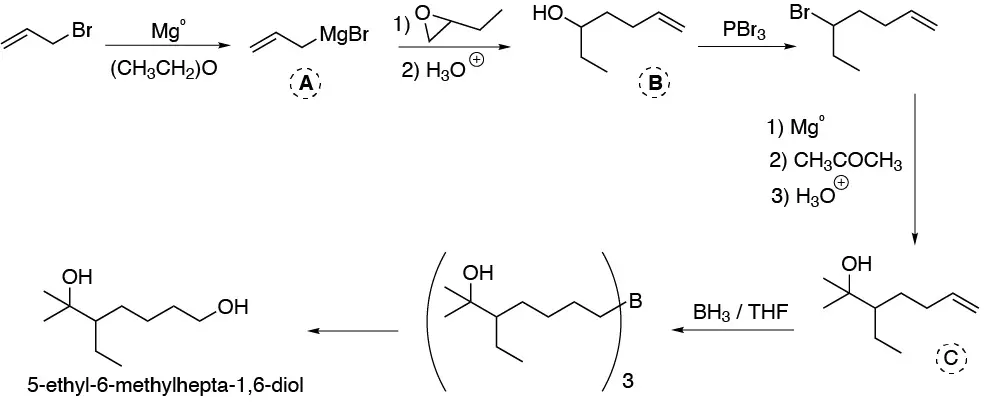
Solution 37:
The key to the proposed synthesis lies in keeping the ketone group at 6 throughout the synthetic sequence. For which, the most useful procedure is to form the cyclic ketal. The rest of the reactions will consist of the reduction of the ester to alcohol with hydride and the esterification of the alcohol with benzoyl chloride. To finish with hydrolysis of the ketal:
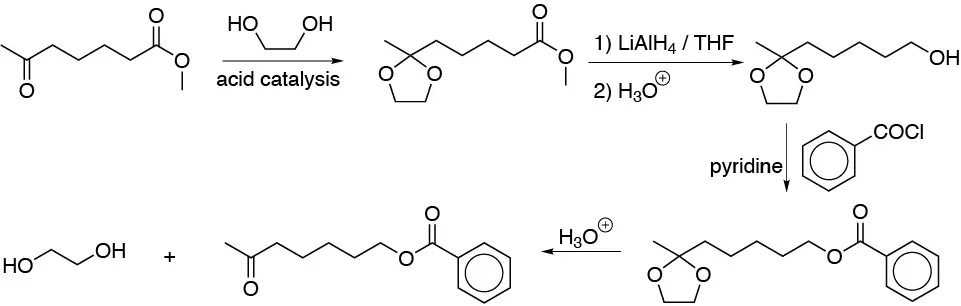
Solution 38:
Compound A will be bromoacetic acid as a result of a Hell-Volhard-Zelinsky reaction. Treatment of it with NaCN will give cyanoacetic acid B. Reaction of B with mineral acid in ethanol will produce both hydrolysis of the nitrile and esterification of the diacid resulting from such hydrolysis, then C will be ethyl malonate (diethyl diethylpropanedioate). If C is treated with base and an alkyl halide will result in alkylation of malonate D. Hydrolysis of this would lead to a β-diacid which undergoes decarboxylation giving 3-phenylpropanoic acid (E):
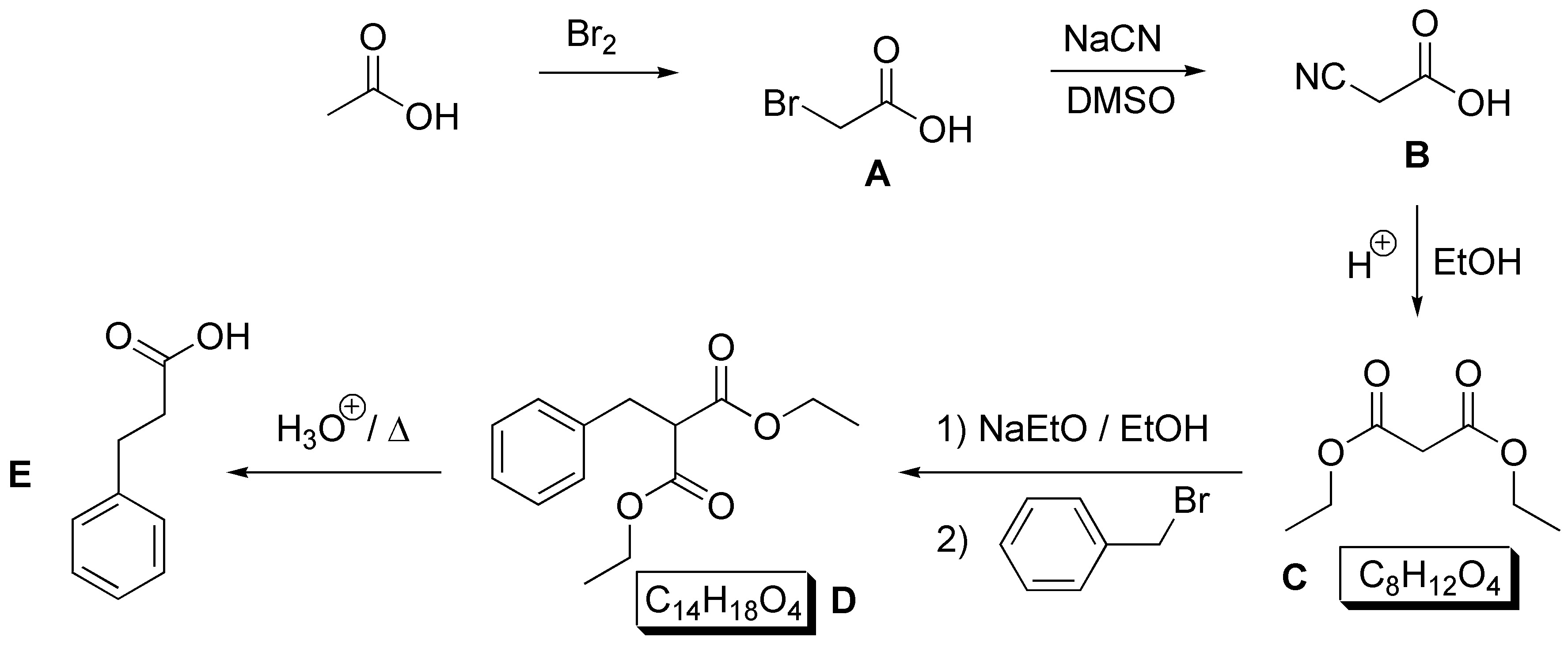
Solution 39:
The structure of B will be that of the epoxide of cyclohexene. This epoxide is nucleophilically opened by an organocuprate giving trans-2-methylcyclohexanol as a mixture of enantiomers. To guess C we must note that hydration of an alkene with anti-Markovnikov regioselectivity forms it, so C must be 1-methylcyclohexene:
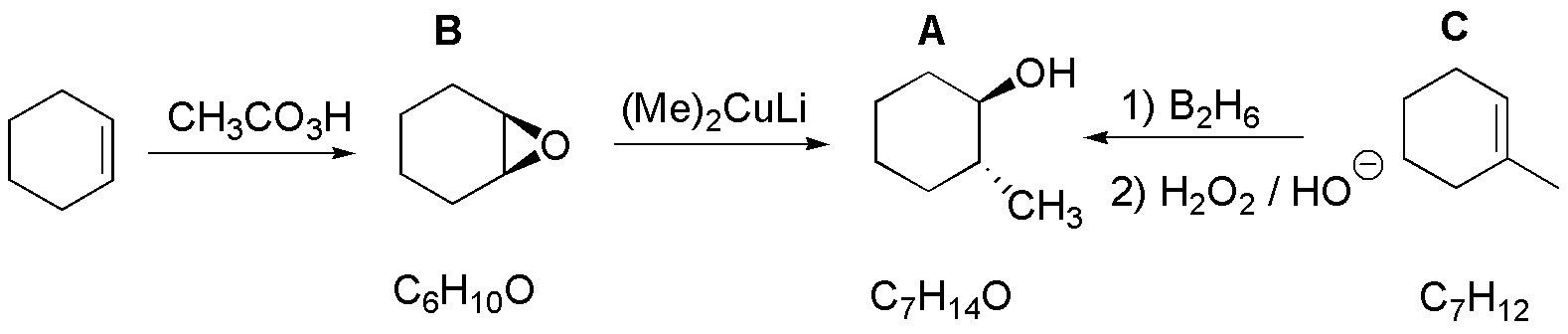
Solution 40:
A is the magnesian of bromobenzene, this reacts with CO2 giving benzoic acid (B), the esterification of which yields methyl benzoate (C). When an ester is treated with a magnesian, it is transformed into a tertiary alcohol in this case triphenylcarbinol (triphenylmethanol) D:

Solution 41:
The purpose of this problem is to try to discriminate primary from secondary alcohols according to their reactivity. When 2,5-hexanediol is treated with a tert-butyldimethylsilyl chloride only the primary is protected as a silylether, which allows oxidation of the secondary alcohol to ketone, subsequent deprotection of the silylether with tetrabutylammonium fluoride gives us the alcohol sought:
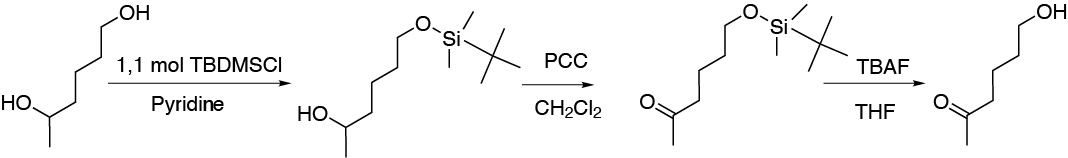
Solution 42:
When an alkene is treated with aqueous mineral acid it produces hydration of the alkene. Subsequently, A is treated with cyclopentanol, whose oxidation will give cyclopentanone B, treating this with a peroxyacid will produce Baeyer-Villiger oxidation giving the corresponding lactone C.
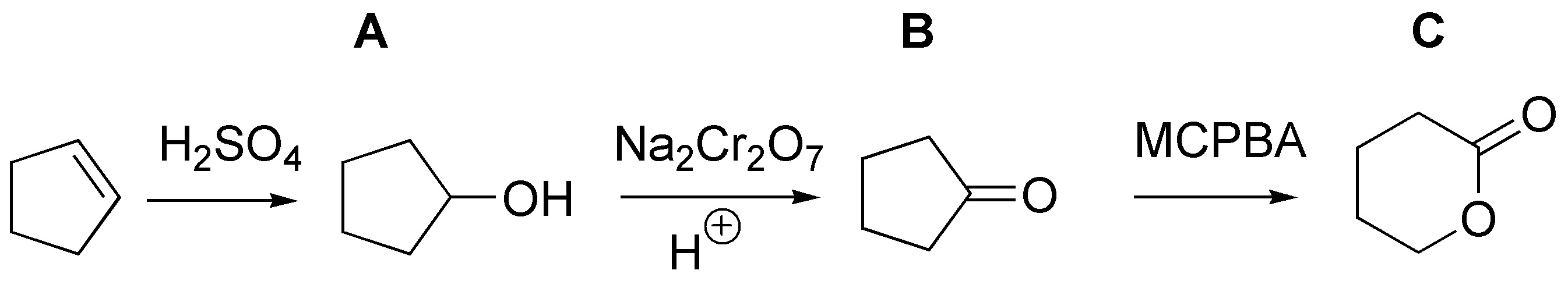
Solution 43:
a) Alkylation of the active methylene compound, ethyl acetylacetate (ethyl 3-oxobutyrate), with propyl bromide gives the corresponding ketoester. By saponification and heating in acid medium it undergoes decarboxylation giving 2-hexanone.
b) It is a cyclic β-dicetone. Therefore, the critical step in the synthesis will be the formation of a cyclic product by condensation: Robinson annulation or similar, so we will need an unsaturated ketone and an enolizable compound. The aldol condensation of propanone (acetone) yields 4-methyl-3-en-pentan-2-one, which can undergo a Michael-type addition of the enolate of diethyl malonate and subsequently undergoes an aldol condensation giving the cyclohexane dione indicated in the scheme. Saponification of said product and subsequent acidification produces the decarboxylation of the resulting ketoacid giving the desired product.
c) Michael-type addition of the enolate of acetophenone (methylphenylketone) onto 3-methylbut-3-en-2-one yields the desired product directly.
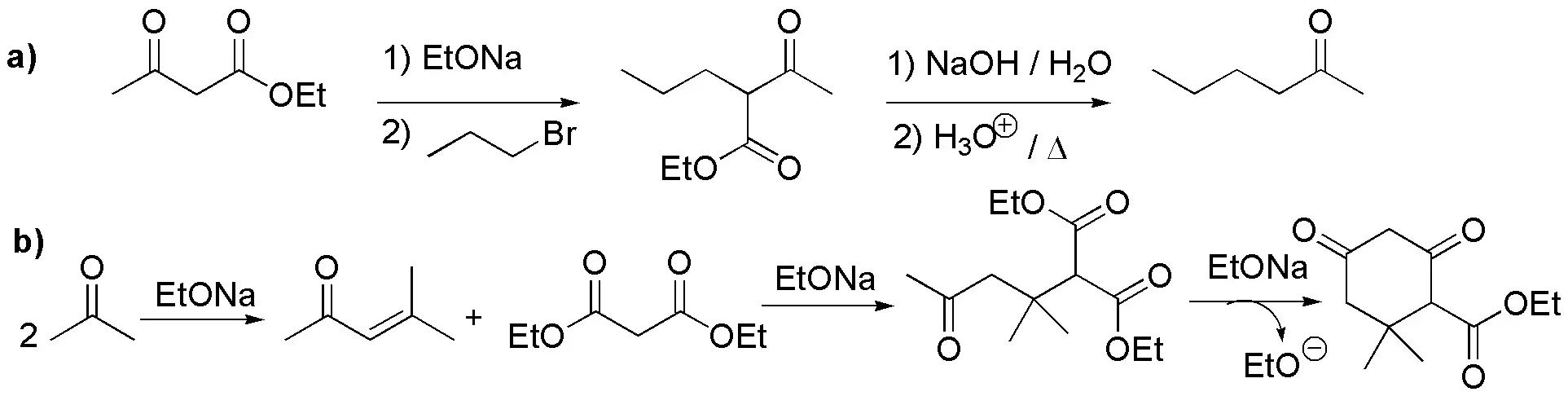
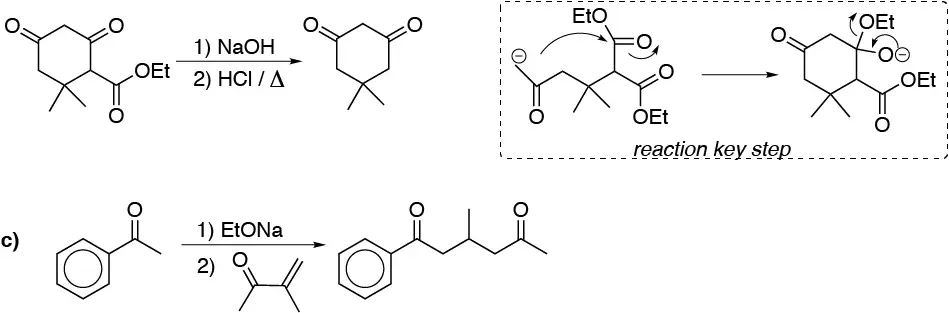
Solution 44:
The indicated synthesis begins with the formation of the corresponding magnesium A, which will nucleophilically attack ethylene oxide producing the alcohol B which is transformed into the chloroalkane C. When this reacts with an acetylide of alkyne D, the indicated alkyne is produced. Hydrogenation with Lindlar catalyst gives the corresponding Z-alkene E, whose epoxidation generates us the disparlure, as a mixture of enantiomers.
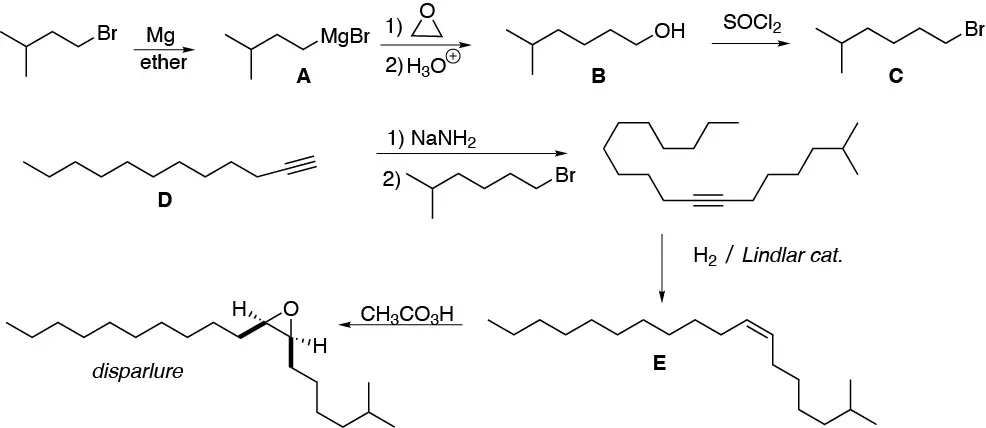
Solution 45:
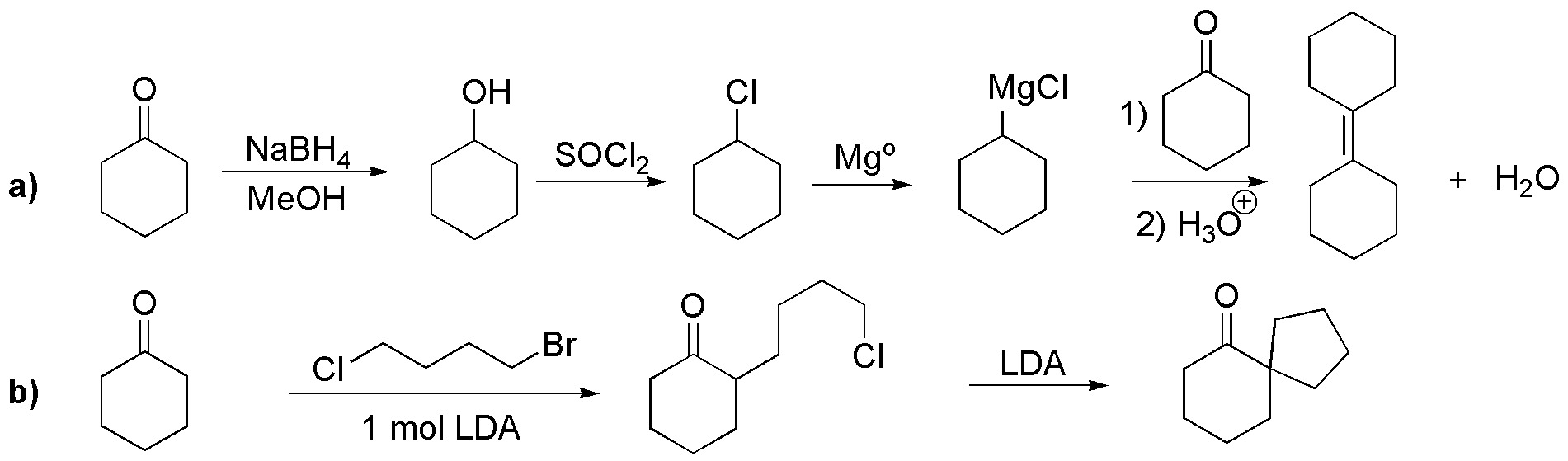
a) cyclohexanone can be readily converted to the magnesian of chlorocyclohexane as shown in the scheme. Addition of such a magnesian to cyclohexanone will produce a dehydrating alcohol giving the desired product, since it is the more stable (more substituted) alkene.
b) A double alkylation must occur at the α carbon with respect to the carbonyl. We take advantage of the different capacity as a leaving group of the bromide and chloride ions, and by treatment with LDA (lithium diisopropylamidide) we will obtain the desired product.
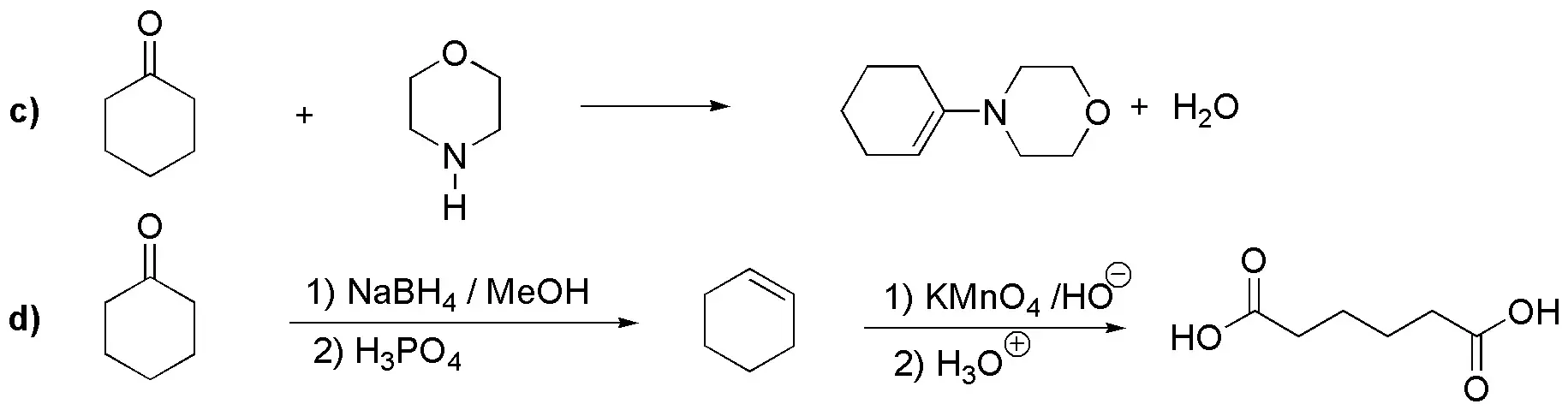
c) It is an enamine as a result of the reaction of cyclohexanone with morpholine (a secondary amine).
d) Cyclohexanone can be converted to cyclohexene by a reduction-dehydration sequence. Oxidation with ozone, or with permanganate in acidic medium, gives us hexanedioic acid.
Solution 46:
The proposed synthesis will consist of a reduction of the nitrile with LiAlH4 to the corresponding primary amine which we subject to dialkylation with bromopropane. There is a risk of peralkylation.
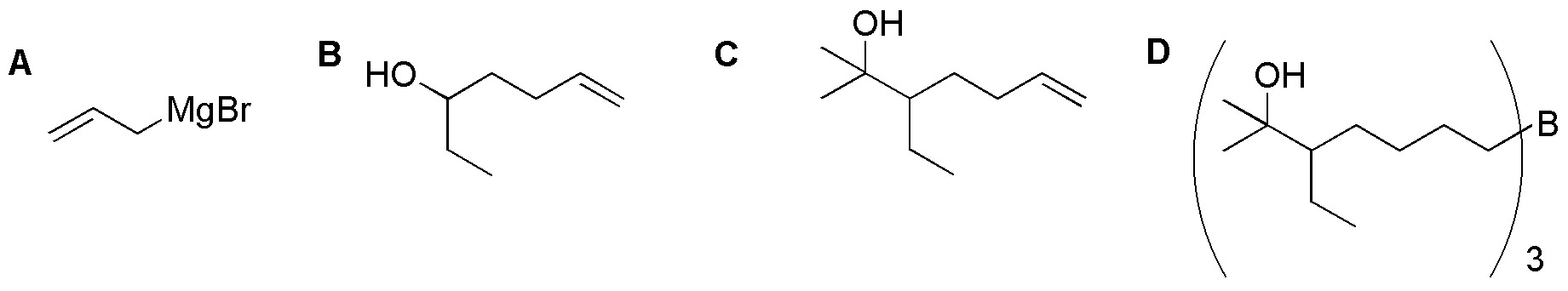
Solution 47:
a) Being a symmetric acyclic product we cleave cyclopentene with ozone giving us the pentanodial and this we subject it to a double Wittig reaction with the indicated phosphorus ylide giving the desired diene.
b) We proceed to the formation of cyclopentanediene from cyclopentene by allylic halogenation and subsequent dehydrohalogenation. The Diels-Alder reaction of this with maleic acid ((Z)-butenodioic acid) gives the bicyclopentanediene diacid
Solution 48:
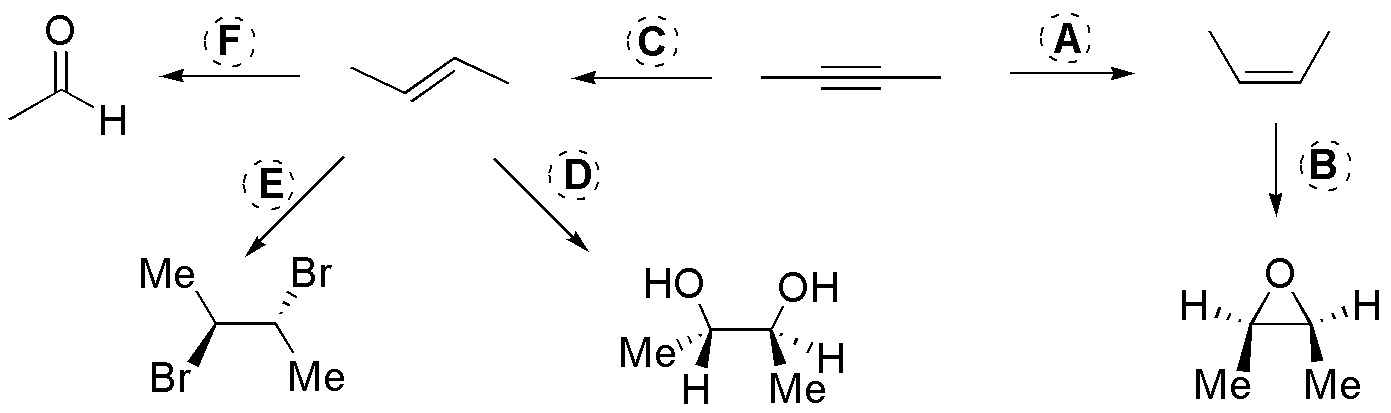
A: It must be H2 / Lindlar cat., to produce the Z-alkene; B: Being an epoxide the resulting product must be a peracid, for example peracetic acid; C: To produce the E-alkene would be a hydrogenation with Na / NH3(l); D: As a diol is formed without we would obtain it with OsO4 / H2O2; E: The dibromide would be obtained by bromination of the alkene, then it is: Br2/Cl4C; F: As the product is acetaldehyde the easiest way would be by ozonolysis: O3 and SMe2.
Full Professor of Organic Chemistry at the University of Granada, with a long-standing research career in Computational Chemistry and molecular modeling and design.